GRETTA:挖掘基因相互作用和协同必需网络的好包!
 今天小果给大家介绍一个2023年6月1日发表于bioinformatics上新鲜出炉的好包——GRETTA!
今天小果给大家介绍一个2023年6月1日发表于bioinformatics上新鲜出炉的好包——GRETTA!

在生物信息学领域,研究基因与基因之间的相互作用以及基因的功能至关重要。然而,通过体外和体内实验来揭示这些网络往往需要大量的资源,限制了样本分析的通量。为了解决这个问题,Yuka Takemon和Marco A Marra开发了一个名为GRETTA的R包,使得利用公开数据进行基因相互作用和协同必需网络分析变得更加简单易行。现在,小果将为大家介绍这个强大的工具。

安装
install.packages(c("devtools", "dplyr","forcats","ggplot2"))devtools::install_github("ytakemon/GRETTA")
公众号后台回复“111”领取本篇代码、基因集或示例数据等文件;
文件编号:231207
果粉福利:生信人必备神器——服务器
平时生信分析学习中有要的小伙伴可以联系小果租赁,粉丝福利都是市场超低价格,赶快找小果领取免费的试用账号吧!
工作流程
基因相互作用
1.安装“GRETTA”并下载附带的数据。
2.选择基因感兴趣的突变细胞系和对照细胞系。
–可以用于根据疾病类型,疾病亚型或氨基酸变化选择细胞系。
3.确定突变细胞系和对照细胞系组之间的差异表达。
–(可选但推荐)。
4.进行in silico基因筛查。
5.可视化结果。
协同必需(co-essential)网络
1.安装“GRETTA”并下载附带的资料。
2.运行相关性系数分析。
–可以用于在特定疾病类型的细胞系中进行的分析。
3.计算负/正曲线的拐点以确定阈值。
4.应用阈值。
5.可视化结果。

一个例子:鉴定ARID1A基因相互作用
ARID1A编码染色质重塑SWItch/非发酵糖(SWI/SNF)复合物的一个成员,并且是癌症中频繁发生突变的基因。已知ARID1A及其同源物ARID1B是彼此的合成致死基因:ARID1A和其同源物ARID1B在细胞中的双重丧失是致死的;然而,单独丧失任何一个基因都不是致死的(Helming等,2014)。这个例子将展示如何使用GRETTA鉴定ARID1A的合成致死互作物并预测这一已知的相互作用。
在本例中,你需要调用以下库。如果这些库尚未安装,请使用install.packages()(例如install.packages(“dplyr”))。
# Load librarylibrary(tidyverse)#> ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──#> ✔ ggplot2 3.4.1 ✔ purrr 1.0.1#> ✔ tibble 3.2.1 ✔ dplyr 1.1.1#> ✔ tidyr 1.3.0 ✔ stringr 1.5.0#> ✔ readr 2.1.4 ✔ forcats 1.0.0#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──#> ✖ dplyr::filter() masks stats::filter()#> ✖ dplyr::lag() masks stats::lag()library(GRETTA)#>#> _______ .______ _______ .___________.___________. ___#> / _____|| _ | ____|| | | /#> | | __ | |_) | | |__ `---| |----`---| |----` / ^#> | | |_ | | / | __| | | | | / /_#> | |__| | | | ----.| |____ | | | | / _____#> ______| | _| `._____||_______| |__| |__| /__/ __#>#> Welcome to GRETTA! The version loaded is: 0.99.2#> The latest DepMap dataset accompanying this package is v22Q2.#> Please refer to our tutorial on GitHub for loading DepMap data and details: https://github.com/ytakemon/GRETTA
下载示例数据
为本教程创建了一个小型数据集,可以使用以下代码下载。
path <- getwd()download_example_data(path)#> Data saved to: /projects/marralab/ytakemon_prj/DepMap/GRETTA/GRETTA_example/
然后,分配变量指向存储 .rda 文件的位置和结果文件应该存储的位置。
gretta_data_dir <- paste0(path,"/GRETTA_example/")gretta_output_dir <- paste0(path,"/GRETTA_example_output/")
探索细胞系
可以通过DepMap的门户网站来探索可用的细胞系,门户网站地址为:https://depmap.org/portal/。然而,在GRETTA中还有一些简单内建的功能可以供用户使用一系列list_available函数,包括list_mutations(), list_cancer_types(), list_cancer_subtypes()。
目前默认使用的DepMap数据版本是22Q2,包含1771个癌细胞系的全基因组测序或全外显子测序注释(1406个细胞系具有RNA-seq数据,375个细胞系具有定量蛋白质组学数据,1086个细胞系具有CRISPR-Cas9敲除筛选数据)。
## Find ARID1A hotspot mutations detected in all cell lineslist_mutations(gene = "ARID1A", is_hotspot = TRUE, data_dir = gretta_data_dir)## List all available cancer typeslist_cancer_types(data_dir = gretta_data_dir)#> [1] "Kidney Cancer" "Leukemia"#> [3] "Lung Cancer" "Non-Cancerous"#> [5] "Sarcoma" "Lymphoma"#> [7] "Colon/Colorectal Cancer" "Pancreatic Cancer"#> [9] "Gastric Cancer" "Rhabdoid"#> [11] "Endometrial/Uterine Cancer" "Esophageal Cancer"#> [13] "Breast Cancer" "Brain Cancer"#> [15] "Ovarian Cancer" "Bone Cancer"#> [17] "Myeloma" "Head and Neck Cancer"#> [19] "Bladder Cancer" "Skin Cancer"#> [21] "Bile Duct Cancer" "Prostate Cancer"#> [23] "Cervical Cancer" "Thyroid Cancer"#> [25] "Neuroblastoma" "Eye Cancer"#> [27] "Liposarcoma" "Gallbladder Cancer"#> [29] "Teratoma" "Unknown"#> [31] "Liver Cancer" "Adrenal Cancer"#> [33] "Embryonal Cancer"## List all available cancer subtypeslist_cancer_subtypes(input_disease = "Lung Cancer", data_dir = gretta_data_dir)#> [1] "Non-Small Cell Lung Cancer (NSCLC), Adenocarcinoma"#> [2] "Small Cell Lung Cancer (SCLC)"#> [3] "Non-Small Cell Lung Cancer (NSCLC), Squamous Cell Carcinoma"#> [4] "Mesothelioma"#> [5] "Non-Small Cell Lung Cancer (NSCLC), Large Cell Carcinoma"#> [6] NA#> [7] "Non-Small Cell Lung Cancer (NSCLC), unspecified"#> [8] "Non-Small Cell Lung Cancer (NSCLC), Adenosquamous Carcinoma"#> [9] "Carcinoid"#> [10] "Non-Small Cell Lung Cancer (NSCLC), Mucoepidermoid Carcinoma"#> [11] "Carcinoma"
选择突变细胞系和对照细胞系组
默认情况下,select_cell_lines()将根据指定基因的功能丧失改变来识别癌细胞系,并将它们分为六组不同的组。
功能丧失性改变包括被注释为“Nonsense_Mutation”(无义突变)、“Frame_Shift_Ins”(框架插入)、“Splice_Site”(剪接位点)、“De_novo_Start_OutOfFrame”(非框架启动子)、“Frame_Shift_Del”(框架删除)、“Start_Codon_SNP”(启动子密码子单倍型)、“Start_Codon_Del”(启动子密码子删除)和“Start_Codon_Ins”(启动子密码子插入)的变异。同时,复制数改变也被纳入考虑,并分为“Deep_del”(深度缺失)、“Loss”(缺失)、“Neutral”(中立)、和“Amplified”(扩增)。
默认分配的细胞系组是:
•Control细胞系不携带具有中性拷贝数(CN)的任何单核苷酸变异(SNVs)或插入和缺失(InDels)。
•HomDel细胞系含有一个或多个纯合有害SNVs,或者具有深度拷贝数损失。
•T-HetDel细胞系含有两个或更多具有中性或拷贝数损失的有害SNVs/InDels杂合子。
•HetDel细胞系含有一个具有中性拷贝数的有害SNVs/InDels杂合子,或者没有拷贝数损失的SNVs/InDels。
•Amplified细胞系不含有拷贝数增加的有害SNVs/InDels。
•Others细胞系含有有害SNVs,其拷贝数增加。
ARID1A_groups <- select_cell_lines(input_gene = "ARID1A", data_dir = gretta_data_dir)#> Selecting mutant groups for: ARID1A in all cancer cell lines## Show number of cell lines in each groupcount(ARID1A_groups, Group)#> # A tibble: 5 × 2#> Group n#>#> 1 ARID1A_HetDel 61#> 2 ARID1A_HomDel 23#> 3 ARID1A_T-HetDel 30#> 4 Control 906#> 5 Others 66
可选的细胞系过滤器
还有几个额外的过滤器可以组合在一起,以缩小我们的搜索范围。这些
•input_aa_change – 通过氨基酸变化(例如“p.Q515*”)。
•input_disease – 通过疾病类型(例如“胰腺癌”)
•input_disease_subtype – 通过疾病亚型(例如“导管腺鳞状细胞癌”)
## Find pancreatic cancer cell lines with ARID1A mutationsARID1A_pancr_groups <- select_cell_lines(input_gene = "ARID1A",input_disease = "Pancreatic Cancer",data_dir = gretta_data_dir)#> Selecting mutant groups for: ARID1A in Pancreatic Cancer, cell lines## Show number of cell lines in each groupcount(ARID1A_pancr_groups, Group)#> # A tibble: 4 × 2#> Group n#>#> 1 ARID1A_HetDel 5#> 2 ARID1A_HomDel 4#> 3 Control 36#> 4 Others 2
检测差异表达
在三种突变癌细胞系组 ARID1A_HomDel、ARID1A_T-HetDel 和 ARID1A_HetDel 中,具有 ARID1A_HomDel 突变的癌细胞系最可能导致 ARID1A 的丢失或表达减少。因此,我们想要检查 ARID1A_HomDel 突变组中的细胞系与对照细胞系相比是否具有显着较少的 ARID1A RNA 或蛋白质表达。
## Select only HomDel and Control cell linesARID1A_groups_subset <- ARID1A_groups %>% filter(Group %in% c("ARID1A_HomDel", "Control"))## Get RNA expressionARID1A_rna_expr <- extract_rna(input_samples = ARID1A_groups_subset$DepMap_ID,input_genes = "ARID1A",data_dir = gretta_data_dir)#> Following sample did not contain RNA data: ACH-000047, ACH-000426, ACH-000658, ACH-000979, ACH-001039, ACH-001063, ACH-001065, ACH-001107, ACH-001126, ACH-001137, ACH-001205, ACH-001212, ACH-001227, ACH-001331, ACH-001544, ACH-001606, ACH-001639, ACH-001675, ACH-001955, ACH-001956, ACH-001957, ACH-002083, ACH-002106, ACH-002109, ACH-002110, ACH-002114, ACH-002116, ACH-002119, ACH-002140, ACH-002141, ACH-002143, ACH-002150, ACH-002156, ACH-002160, ACH-002161, ACH-002179, ACH-002181, ACH-002186, ACH-002189, ACH-002198, ACH-002202, ACH-002210, ACH-002212, ACH-002217, ACH-002228, ACH-002229, ACH-002230, ACH-002233, ACH-002234, ACH-002239, ACH-002243, ACH-002247, ACH-002249, ACH-002250, ACH-002257, ACH-002261, ACH-002263, ACH-002265, ACH-002269, ACH-002278, ACH-002280, ACH-002282, ACH-002283, ACH-002284, ACH-002285, ACH-002294, ACH-002295, ACH-002296, ACH-002297, ACH-002298, ACH-002304, ACH-002305, ACH-002399, ACH-002874, ACH-002875
并非所有细胞系都包含RNA和/或蛋白质表达谱,并且并非所有蛋白质都通过质谱检测到。(有关数据生成的详细信息,请参见DepMap网站。)
## Get protein expressionARID1A_prot_expr <- extract_prot(input_samples = ARID1A_groups_subset$DepMap_ID,input_genes = "ARID1A",data_dir = gretta_data_dir)## Produces an error message since ARID1A protein data is not available
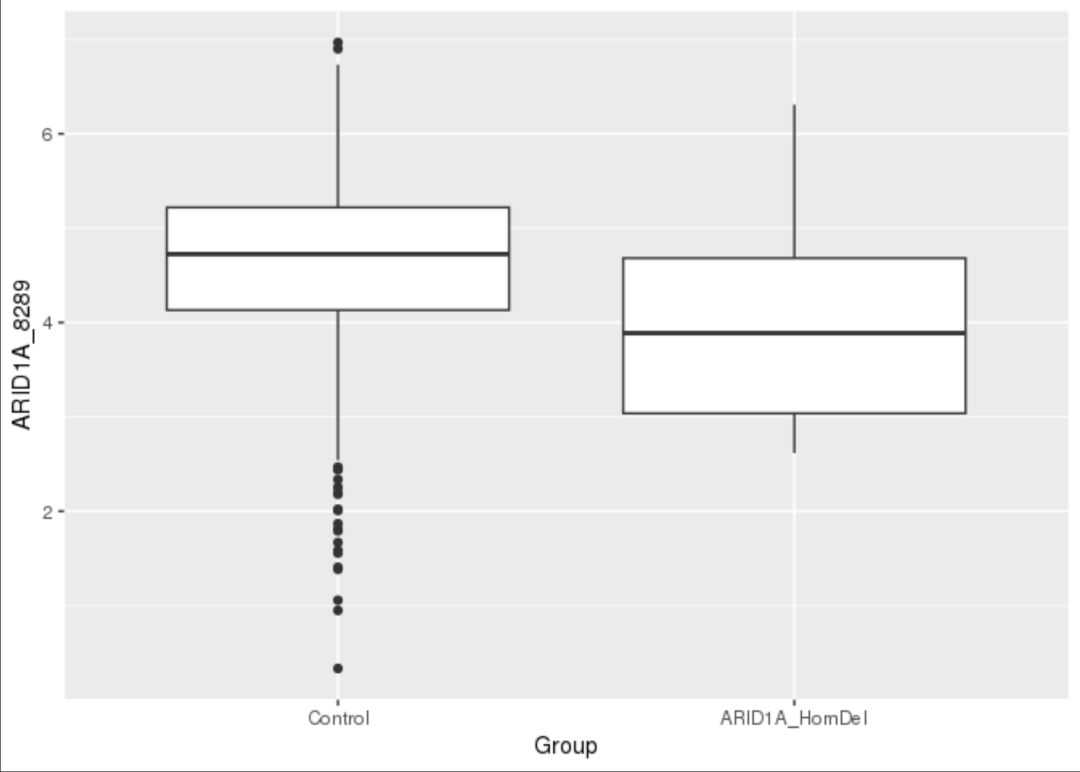
使用Welch的t检验,我们可以检查相对于对照细胞系,ARID1A_HomDel细胞系中的ARID1A RNA表达量(以TPM为单位)是否显著降低。
## Append groups and test differential expressionARID1A_rna_expr <- left_join(ARID1A_rna_expr,ARID1A_groups_subset %>% select(DepMap_ID, Group)) %>%mutate(Group = fct_relevel(Group,"Control")) # show Control group first#> Joining with `by = join_by(DepMap_ID)`## T-testt.test(ARID1A_8289 ~ Group, ARID1A_rna_expr)#>#> Welch Two Sample t-test#>#> data: ARID1A_8289 by Group#> t = 2.5764, df = 22.873, p-value = 0.01692#> alternative hypothesis: true difference in means between group Control and group ARID1A_HomDel is not equal to 0#> 95 percent confidence interval:#> 0.1146094 1.0498810#> sample estimates:#> mean in group Control mean in group ARID1A_HomDel#> 4.635784 4.053539## plotggplot(ARID1A_rna_expr, aes(x = Group, y = ARID1A_8289)) +geom_boxplot()
进行全基因组in silico遗传筛选
在确定ARID1A_HomDel组中的细胞系与Control细胞系相比在RNA表达上具有统计学上显著降低之后,下一步是使用screen_results()执行in silico基因筛选。这使用DepMap的全基因组CRISPR-Cas9敲除筛选中产生的依赖概率(或“致死概率”)。
致死概率(Lethality probabilities)的范围从0.0到1.0,每个被筛查的癌细胞系中每个基因敲除(共针对739个癌细胞系中的18,334个基因)的致死概率都有所量化。一个基因敲除的致死概率为0.0表明该基因对于该细胞系是非致命的,而一个基因敲除的致死概率为1.0表明该基因是致命的(即非常致命)。更多详细信息可参见Meyers, R., et al., 2017。
screen_results()函数的核心进行了多个Mann-Whitney U检验,比较了突变组和对照组中每个靶基因的致死概率。生成的数据框具有以下列:
•GeneName_ID:Hugo symbol带有NCBI基因ID
•GeneNames:Hugo symbol
•median,mean,sd,iqr:控制和突变组依赖概率的中位数、均值、标准差(sd)和四分位数范围(iqr)。依赖概率的范围从零到一,其中一表示必需基因(即基因敲除是致死的),零表示非必需基因(基因敲除不是致死的)
•Pval:控制和突变组之间的Mann-Whitney U检验的P值。
•Adj_pval:经过BH校正的P值。
•log2FC_by_median:以log2为基准归一化的依赖概率中位数变化倍数(突变/对照)。
•log2FC_by_mean:以log2为基准归一化的依赖概率均值变化倍数(突变/对照)。
•CliffDelta:突变和对照依赖概率之间的Cliff’s delta非参数效应大小。范围介于-1到1之间。
•dip_pval:Hartigan的dip测试的p值。测试突变依赖概率的分布是否为单模态。如果dip测试被拒绝(p值<0.05),这表明存在多模态依赖概率分布,并且可能有其他因素导致这种分离。
•Interaction_score:从带符号的p值中得出的组合值:-log10(Pval) * sign(log2FC_by_median)。负分表示致死遗传相互作用,正分表示缓解遗传相互作用。
注意 这个过程可能需要几个小时,具体取决于分配的核心数。小果下面的例子 GI_screen() 花了大约2个小时来处理。为了节省时间,小果已经为您预先处理了这一步。
ARID1A_mutant_id <- ARID1A_groups %>% filter(Group %in% c("ARID1A_HomDel")) %>% pull(DepMap_ID)ARID1A_control_id <- ARID1A_groups %>% filter(Group %in% c("Control")) %>% pull(DepMap_ID)## See warning above.## This can take several hours depending on number of lines/cores used.screen_results <- GI_screen(control_id = ARID1A_control_id,mutant_id = ARID1A_mutant_id,core_num = 5, # depends on how many cores you haveoutput_dir = gretta_output_dir, # Will save your results here as well as in the variabledata_dir = gretta_data_dir,test = FALSE) # use TRUE to run a short test to make sure all will run overnight.## Load prepared ARID1A screen resultload(paste0(gretta_data_dir,"/sample_22Q2_ARID1A_KO_screen.rda"), envir = environment())
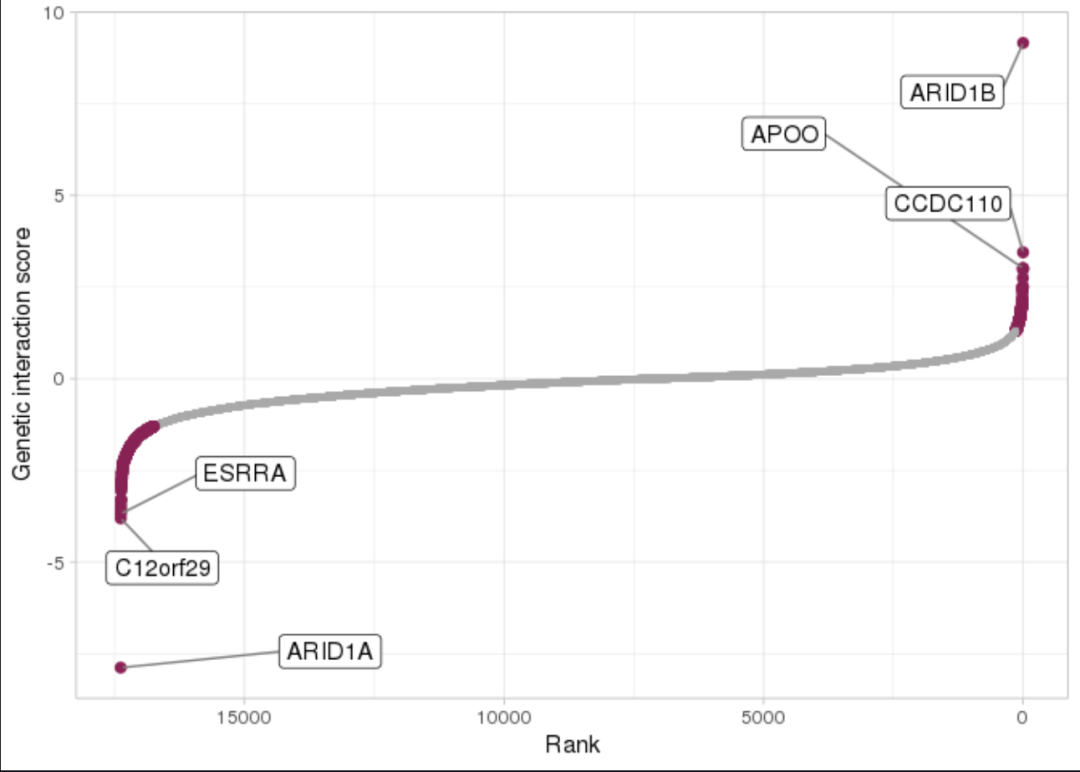
我们可以快速确定GRETTA是否预测了任何致死性遗传相互作用。我们使用Pval截断值为0.05,并根据Interaction_score进行排名。
screen_results %>%filter(Pval < 0.05) %>%arrange(-Interaction_score) %>%select(GeneNames:Mutant_median, Pval, Interaction_score) %>% head#> # A tibble: 6 × 5#> GeneNames Control_median Mutant_median Pval Interaction_score#>#> 1 ARID1B 0.0579 0.515 6.84e-10 9.16#> 2 CCDC110 0.0165 0.0303 3.54e- 4 3.45#> 3 APOO 0.0168 0.0283 9.61e- 4 3.02#> 4 NHS 0.0352 0.0539 9.69e- 4 3.01#> 5 SLC66A2 0.00793 0.0134 1.06e- 3 2.98#> 6 ATXN7L1 0.0138 0.0259 1.78e- 3 2.75
小果可以发现,ARID1B,一种已知的与ARID1A的合成致死相互作用,是此列表的顶部。
可选步骤:进行小规模筛选
要进行小型体内筛选,可以在 gene_list = 参数中提供基因列表。
small_screen_results <- GI_screen(control_id = ARID1A_control_id,mutant_id = ARID1A_mutant_id,gene_list = c("ARID1A", "ARID1B", "SMARCA2", "GAPDH", "SMARCC2"),core_num = 5, # depends on how many cores you haveoutput_dir = gretta_output_dir, # Will save your results here as well as in the variabledata_dir = gretta_data_dir)
可视化筛选结果
最后,一旦完成计算机筛选,就可以使用plot_screen()函数快速可视化结果。阳性遗传相互作用分数表示潜在的合成致死遗传互作物,而负分数表示潜在的缓解遗传互作物。不出所料,小果确定了ARID1B是ARID1A的合成致死相互作用物。
## Visualize results, turn on gene labels,## and label three genes each that are predicted to have## lethal and alleviating genetic interactions, respectivelyplot_screen(result_df = screen_results,label_genes = TRUE,label_n = 3)#> Warning: Removed 7 rows containing missing values (`geom_point()`).
示例2:鉴定ARID1A协同必需(co-essential)基因
作用于相同或协同途径以及相同复合物中的基因,其功能受到扰动后表现出相似的适应效应,这些具有类似效应的基因被视为“共同必需基因”。多个研究使用绘制共同必需基因图谱的策略,为先前注释的基因分配功能以及鉴定大型复合物的新亚基(Wainberg et al. 2021; Pan et al. 2018)。
考虑到ARID1A是哺乳动物SWI/SNF复合物已知的亚基(Mashtalir等2018),小果预计SWI/SNF复合物的成员将与ARID1A共享共同必需性。本示例将展示如何使用GRETTA绘制ARID1A的共必需基因网络。
鉴定具有最高相关性系数的基因
为了确定共必需基因,我们将对ARID1A基因敲除效应与18,333个基因的敲除效应之间进行多个皮尔逊相关系数分析。我们将通过计算排序系数曲线的拐点来确定一个截断值。不出所料,我们发现编码SWI / SNF亚基的基因SMARCE1和SMARCB1是前两个共必需基因。
注意 此过程可能需要几分钟的时间。小果下面的示例 coessential_map() + get_inflection_points() 处理大约需要17分钟的时间。为了节省时间,小果已经为您预先处理了这一步骤。
## Map co-essential genescoess_df <- coessential_map(input_gene = "ARID1A",data_dir = gretta_data_dir,output_dir = gretta_output_dir)## Calculate inflection points of positive and negative curve using co-essential gene results.coess_inflection_df <- get_inflection_points(coess_df)接下来,我们对包含共 essential 网络数据的数据框进行注释并进行可视
化。
## Combine and annotate data frame containing co-essential genescoess_annotated_df <- annotate_coess(coess_df, coess_inflection_df)plot_coess(result_df = coess_annotated_df,inflection_df = coess_inflection_df,label_genes = TRUE, # Should gene names be labeled?label_n = 3) # Number of genes to display from each end
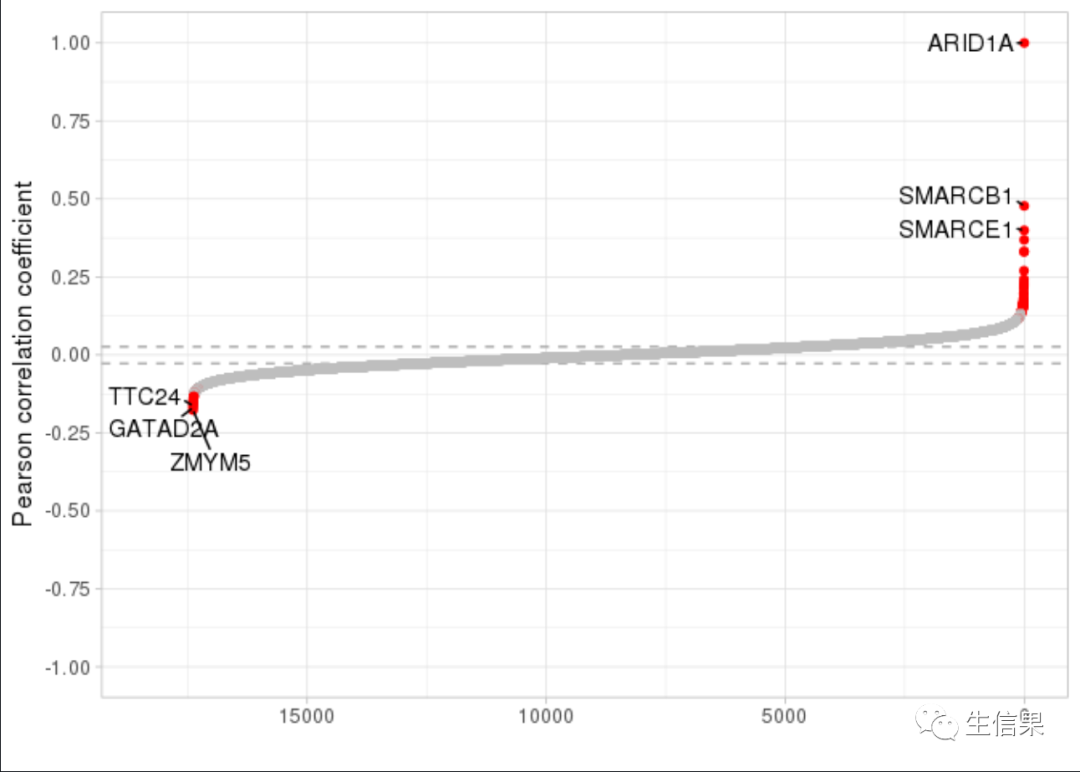
小果还发现,与ARID1A共同至关重要的前十个基因中有八个已知的SWI/SNF复合物亚基,即ARID1A、SMARCB1、SMARCE1、SMARCC1、SS18、DPF2、SMARCC2和SMARCD2。
## Show top 10 co-essential genes.coess_annotated_df %>% arrange(Rank) %>% head(10)#> # A tibble: 10 × 9#> GeneNameID_A GeneNameID_B estimate statistic p.value parameter Rank#>#> 1 ARID1A_8289 ARID1A_8289 1 Inf 0 1086 1#> 2 ARID1A_8289 SMARCB1_6598 0.477 17.9 7.45e-59 1086 2#> 3 ARID1A_8289 SMARCE1_6605 0.399 14.3 4.30e-39 1086 3#> 4 ARID1A_8289 SMARCC1_6599 0.369 13.1 9.35e-33 1086 4#> 5 ARID1A_8289 SS18_6760 0.332 11.6 4.85e-26 1086 5#> 6 ARID1A_8289 DPF2_5977 0.330 11.5 1.15e-25 1086 6#> 7 ARID1A_8289 SMARCD2_6603 0.270 9.22 1.10e-16 1086 7#> 8 ARID1A_8289 SMARCC2_6601 0.242 8.22 2.34e-13 1086 8#> 9 ARID1A_8289 BCL2_596 0.231 7.82 4.05e-12 1086 9#> 10 ARID1A_8289 CBFB_865 0.224 7.58 2.07e-11 1086 10#> # ℹ 2 more variables: Padj_BH, Candidate_gene
特定癌症类型的可选过滤器
用户还可以使用input_disease = “”选项按疾病类型进行子集划分,或使用input_cell_lines = c()选项在预先选定的细胞系组内进行子集划分,而不是在整个可用细胞系中映射重要性。下面小果提供了一个ARID1A必需基因映射用于胰腺癌的示例。
注意 根据子集步骤后可用的细胞系数量,屈曲点计算和阈值可能不是最优的。在解释这些结果时请谨慎使用。
## Map co-essential genes in pancreatic cancers onlycoess_df <- coessential_map(input_gene = "ARID1A",input_disease = "Pancreatic Cancer",core_num = 5, ## Depending on how many cores you have access to, increase this value to shorten processing time.data_dir = gretta_data_dir,output_dir = gretta_output_dir,test = FALSE)
针对自定义细胞系的可选过滤器
我们还可以使用 input_cell_lines = c() 选项,在手动定义的细胞系列表中映射必需性。
注意 根据所提供的细胞系数量,可能不会计算拐点和变化点。在解释这些结果时请谨慎使用。
custom_lines <- c("ACH-000001", "ACH-000002", "ACH-000003",...)coess_df <- coessential_map(input_gene = "ARID1A",input_cell_lines = custom_lines,core_num = 5, ## Depending on how many cores you have access to, increase this value to shorten processing time.data_dir = gretta_data_dir,output_dir = gretta_output_dir,test = FALSE)
结语
你想要分析基因相互作用和协同必需网络吗?试试GRETTA,我们可以利用DepMap的公开数据进行in silico的基因筛选。GRETTA还可以帮助你可视化和解释结果。记得我们上面的例子吗,如何用GRETTA发现ARID1A的基因相互作用和协同必需基因。GRETTA已经有365个访问者和135个安装者,未来可期!
参考文献
[1] Takemon Y, Marra MA. GRETTA: an R package for mapping in silico genetic interaction and essentiality networks. Bioinformatics. 2023 Jun 1;39(6):btad381. doi: 10.1093/bioinformatics/btad381. PMID: 37326978; PMCID: PMC10284671.


往期推荐