小果的单细胞分析保姆级教程!手把手教你从单细胞数据下载和处理到单细胞注释!!
小果的单细胞分析保姆级教程!手把手教你对单细胞数据下载和处理到单细胞注释!!
最近单细胞的数据分析又悄悄的火了起来,小伙伴也了解了不少,但是不知道从何下手,而且呢有关单细胞数据的下载也是一头雾水。其实呢单细胞数据处理也并不是很复杂,只是入门不容易,一旦上手后续的分析就很简单。还有呢,我们在做单细胞转录组数据分析时候,他的难点主要在于细胞的质量不确定,细胞的数量大,从单细胞测序技术诞生至今,测到的细胞通量越来越高。所以小果这里出一期教程可以教小伙伴入门单细胞,让我们从数据下载到处理,小伙伴学会了之后,可以了解单细胞处理的大致流程。下面小果呢,就带小伙伴去下载自己需要的单细胞数据还有如何去处理。
我们一般的对单细胞数据处理呢,肯定是需要和自己做的疾病或者肿瘤相关的单细胞数据集,我们往往用的做的下载方式就是在NCBI这个平台,GEO数据库收集,下面小果就带小伙伴看看如何去下载!
我们一般打开这个网址:https://www.ncbi.nlm.nih.gov/
在这里输入我们的相关的信息搜索,这里小图试验肺癌进行展示,lung cancer 我们在搜索的时候后面要加入单细胞相关的关键字,lung caner scRNAseq 然后在加上 human人类,一般就使用lung caner scRNAseq human 如果是其他疾病 换一下就可以,比如宫颈癌 cervical cancer scRNAseq human
来看看效果
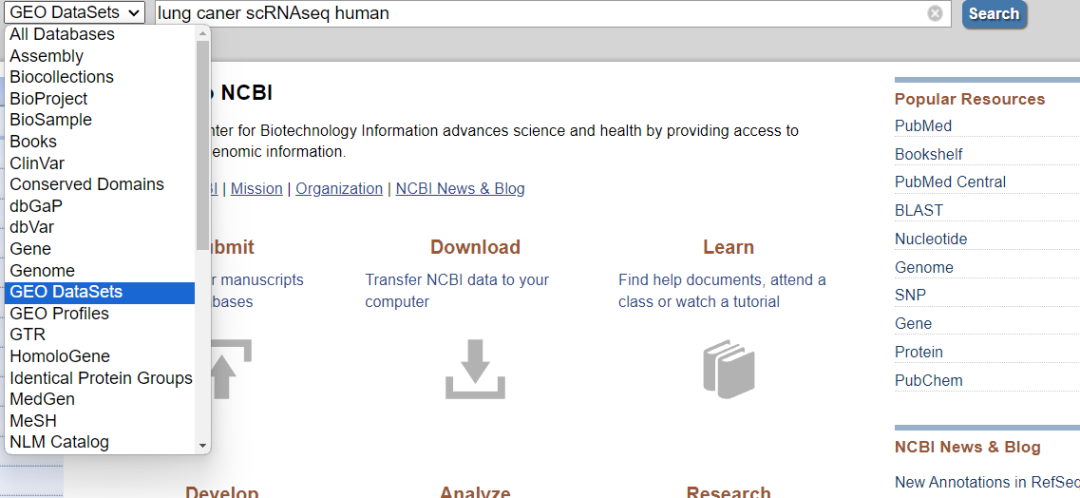
这里需要把搜索来源选择GEO数据库,然后:
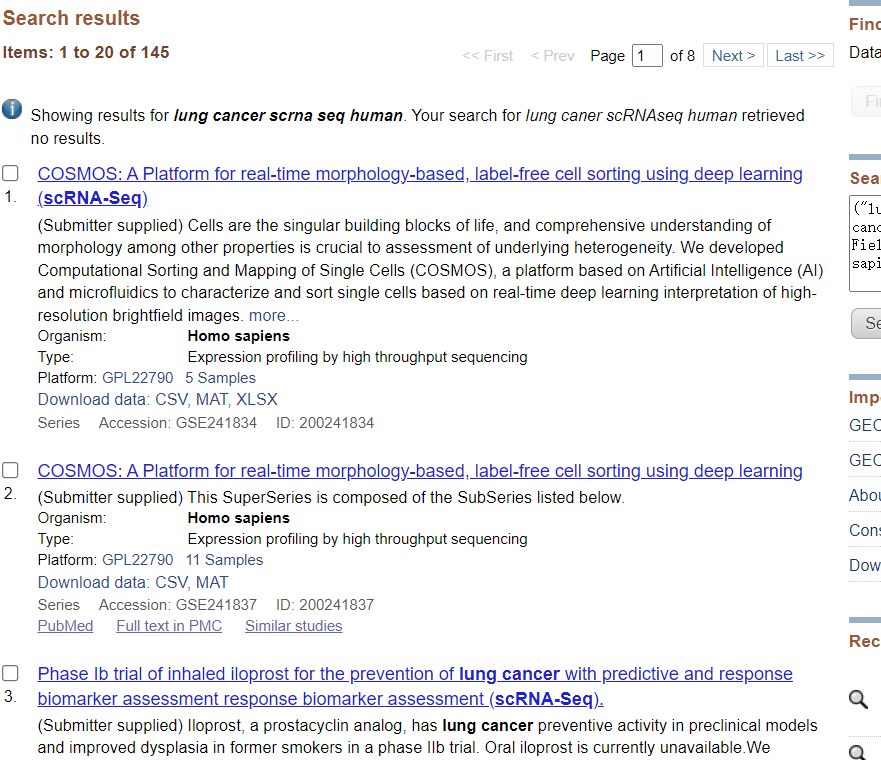
然后就会有下面的界面,这就需要小伙伴去自行去选择,我们随机点击查看一下
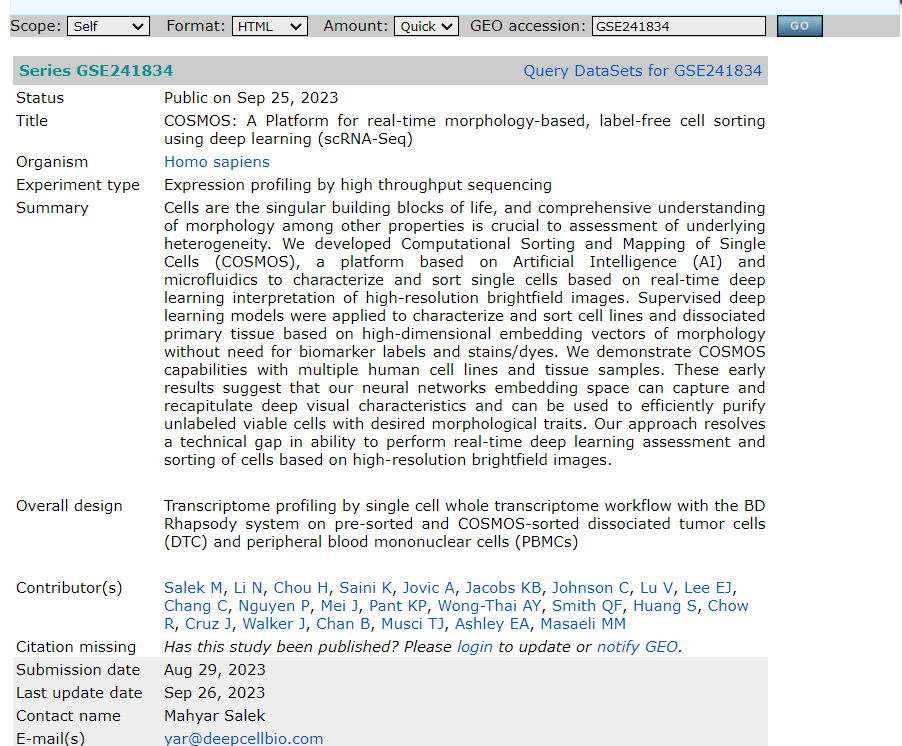
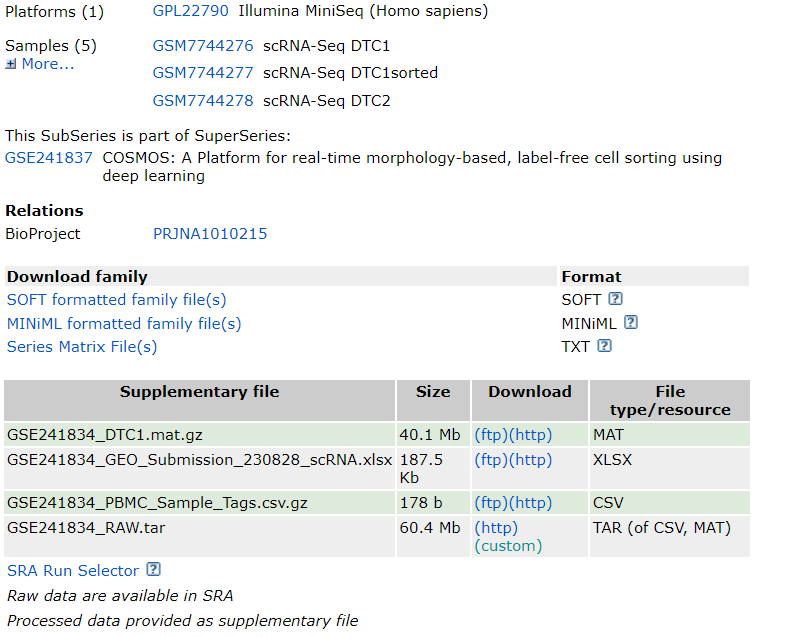
这里有很多单细胞数据,其实我们选择下载的也是有目的的,我们如果选择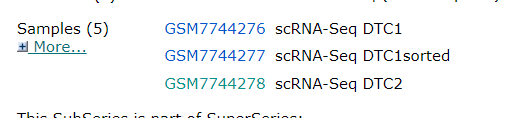
这三个里面的一种,点击进去下载
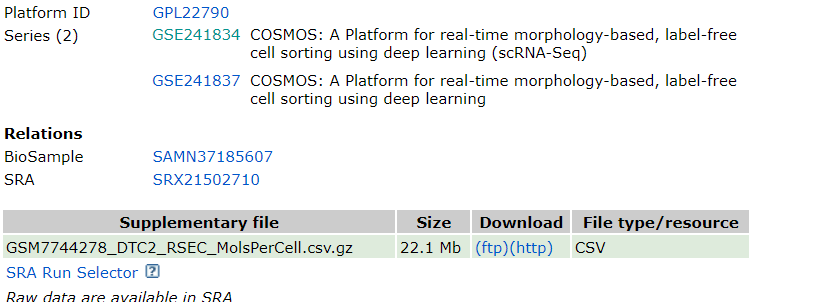

下载这个就可以,这种是CSV格式的,下面小果会教小伙伴导入数据格式的方式,其实我们下载数据集可能还有其他数据格式,一般最多的就是H5格式,还有mtx格式,这里小伙伴不用担心,小果都会教小伙伴,我们来看一个H5格式的,
下载还解压后
就会这样,
还有一种

我们下面对这三个格式导入进行学习,
我们先从H5格式学习,这个比较简单一行代码就学会了,这里小果随便用一个数据集展示,
library(hdf5r)library(Seurat)library(dplyr)
我们还要加载一下这个包,用来处理数据的
data_sample <- Read10X_h5("GSM4983457_NC_filtered_feature_bc_matrix.h5") #导入H5格式数据data_seurat <- CreateSeuratObject(data_sample,project = "data_sample") #后面就可以进行单细胞处理的标准流程了
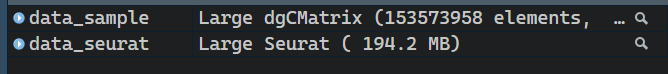
这样就导入成功了,
下面我们对mtx格式进行导入,
而我们往往下载mtx格式的一般会有这三个文件
# 读取10x数据,data.dir参数指定存放文件的路径seurat_data <- Read10X(data.dir = "E:\JYStudio\Rtest\Ztest1\单细胞\单细胞数据处理\Tumor")##这个小伙伴要自行设置存放这个文件的目录,# 然后我们创建Seurat对象data_seurat <- CreateSeuratObject(counts = seurat_data,project = "GSM5155196_352",min.features = 200,min.cells = 3)这个就和上述创建对象,后续就可以进行单细胞的标准流程了使用这个代码就可以导入数据,
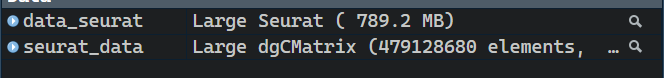
导入后就是一样的,
最后一种就是csv格式的,小伙伴有的下载只有csv格式的怎么办,小果这里教大家
##第二种library(tidyr)library(tidyverse)library(org.Mm.eg.db)library(scater)library(scran)library(pheatmap)library(limma)library(edgeR)library(dynamicTreeCut)library(cluster)library(monocle)aa <- read.csv("GSE122340.csv",row.names = 1,header = T)all.counts <- as.matrix(aa)pbmc <- CreateSeuratObject(counts = all.counts, project = "TCGA", names.delim = "-",min.cells = 3, min.features = 200)
我们把包加载好了之后,然后导入自己csv格式文件,然后导入成功后,就可以正常进行单细胞数据分析了!
好了关于数据下载和导入就完成了,小伙伴快去动手试试,下面我们就开始正式分析了!
这里小果把导入数据处理一下:
# 查看Seurat对象的基本信息data_seurat
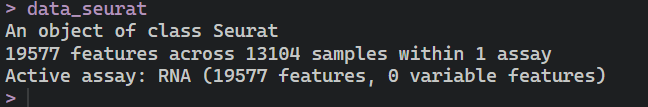
这个就是可以看到我们导入单细胞数据的总体
data_seurat[["percent.mt"]]<-PercentageFeatureSet(data_seurat,pattern = "MT-")VlnPlot(data_seurat,features = c("nCount_RNA","nFeature_RNA","percent.mt"))
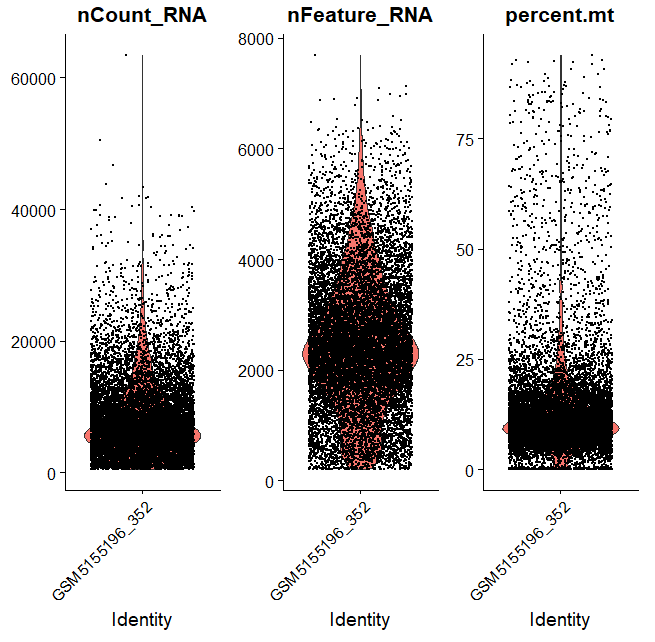
我们以不同形式展示看看基因数据那些细胞,比如线粒体等
然后我们要剔除,
plot1<-FeatureScatter(data_seurat,feature1 = "nCount_RNA",feature2 = "percent.mt")plot2<-FeatureScatter(data_seurat,feature1 = "nCount_RNA",feature2 = "nFeature_RNA")CombinePlots(plots = list(plot1,plot2))
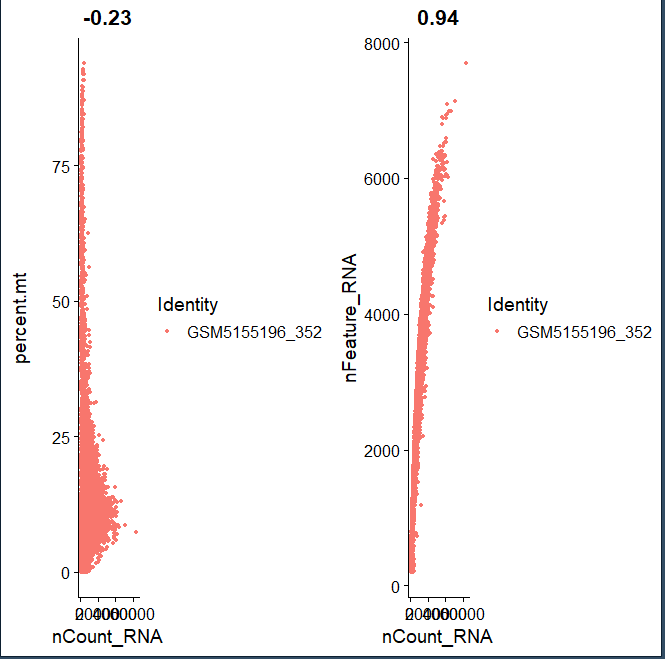
剔除情况,这个无关紧要,
pbmc<-subset(data_seurat,subset = nFeature_RNA>200 & nFeature_RNA<2500 & percent.mt<5)这个根据小伙伴自己的情况去选择,范围3000还有5000 还有最小为200还是多少,小伙伴可以根据自身情况,
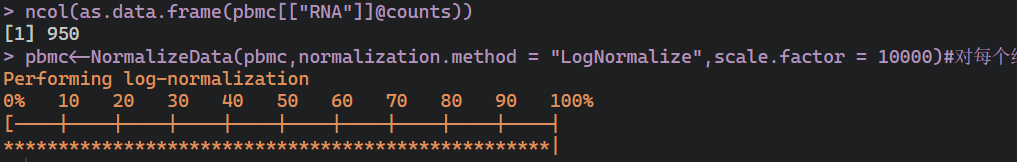
ncol(as.data.frame(pbmc[["RNA"]]@counts))pbmc<-NormalizeData(pbmc,normalization.method = "LogNormalize",scale.factor = 10000)#对每个细胞标准化.
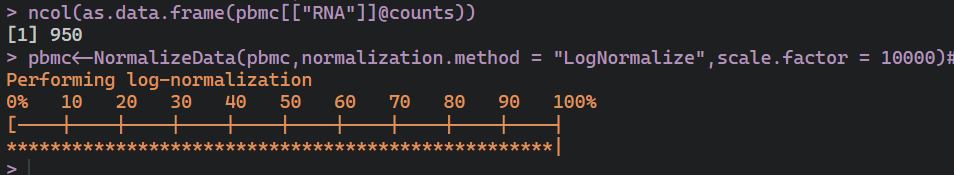
这里我们对每个细胞都进行标准化,
下面我们对数据进行处理
这次小果继续教小伙伴对数据处理,还有我们的重头戏对单细胞注释,下面我们开始吧
上次我们处理到,
对每个细胞都进行了标准化,下面我们就是该寻找高变的基因,这样才能找到与我们整体单细胞相关的基因之间的关系,来看看如何寻找
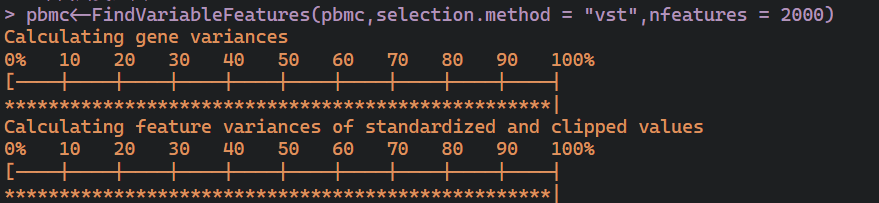
#寻找高变基因pbmc<-FindVariableFeatures(pbmc,selection.method = "vst",nfeatures = 2000)
然后我们进行
#PCAtop10<-head(VariableFeatures(pbmc))
这行代码我们查看一下前十个的高变基因,可以发现我的这个只有6个高变基因,可能我选取的数值比较少,不过不影响

我们继续往下走
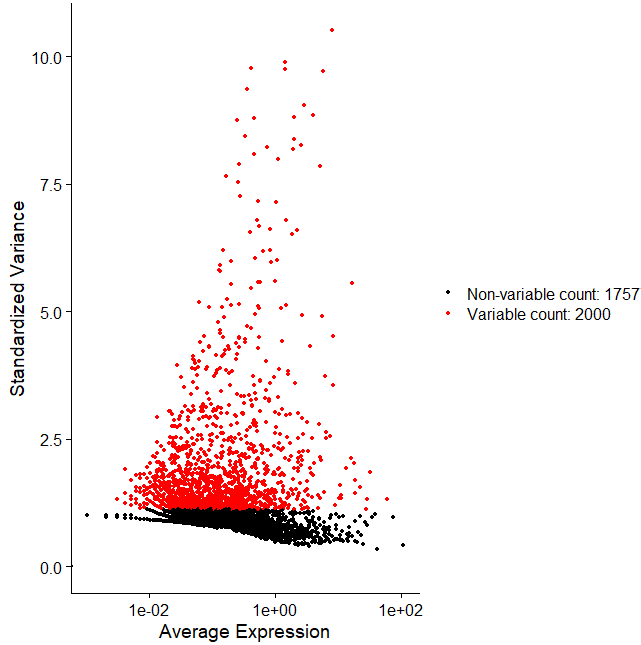
plot1<-VariableFeaturePlot(pbmc)= plot1,points=top10,repel=T)plot1
plot2

把我们找到的高变基因放在火山图上面,我们寻找高变基因也是为了在后续寻找细胞亚群的时候分离有用,我们后续再说,下面我们对基因进行标准化,和上面不同这次是对基因
all.genes<-rownames(pbmc)pbmc<-ScaleData(pbmc,features = all.genes)#每种基因在细胞中的标准化pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")

这里分析好了,我们就可以后续分析,对细胞进行聚类:
pbmc<-RunPCA(pbmc,features = VariableFeatures(object = pbmc))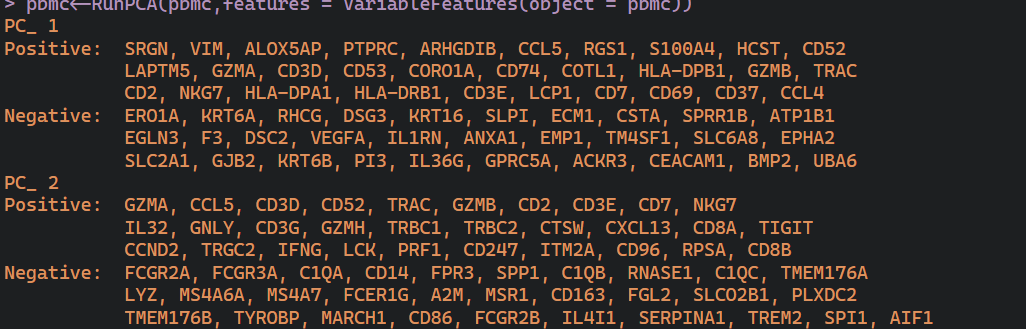
在做一个PCA分析,看看不同类之间的关联
print(pbmc[["pca"]],dims=1:5,nfeatures=5)
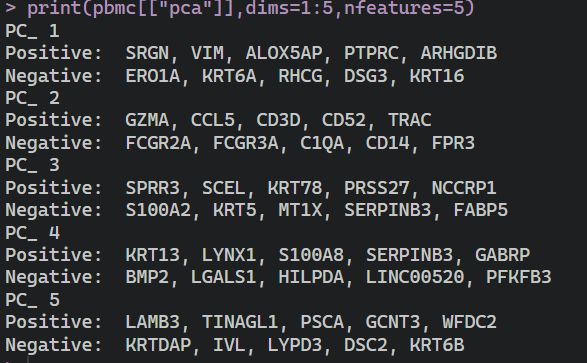
VizDimLoadings(pbmc,dims = 1:2,reduction = "pca")
DimPlot(pbmc,reduction = "pca")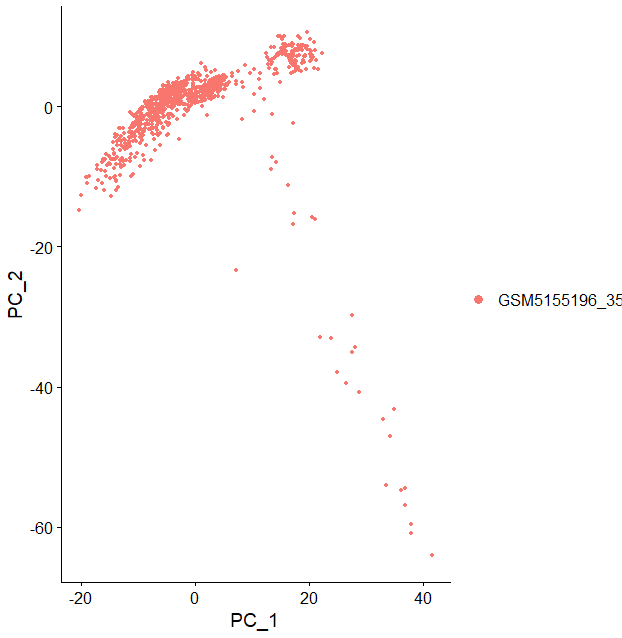
DimHeatmap(pbmc,dims=1,cells=500,balanced = T)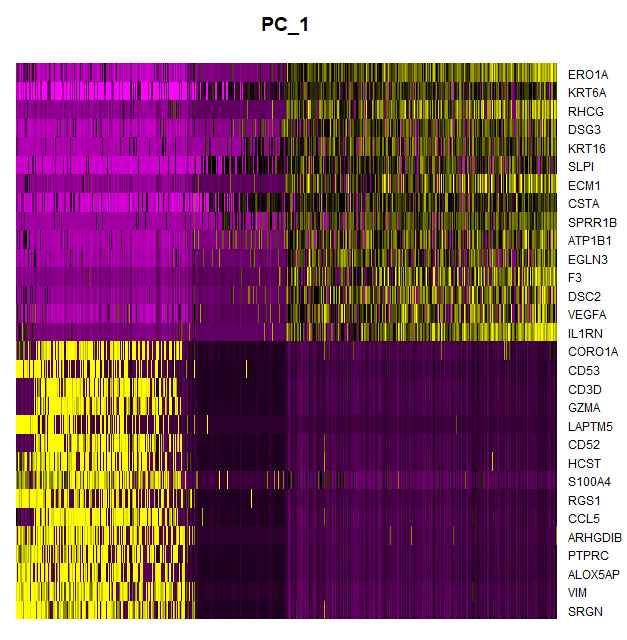
这个就可以看到每个基因在不同的亚群之间的表达如何,还可以分开来看
DimHeatmap(pbmc,dims = 1:15,cells=500,balanced = T)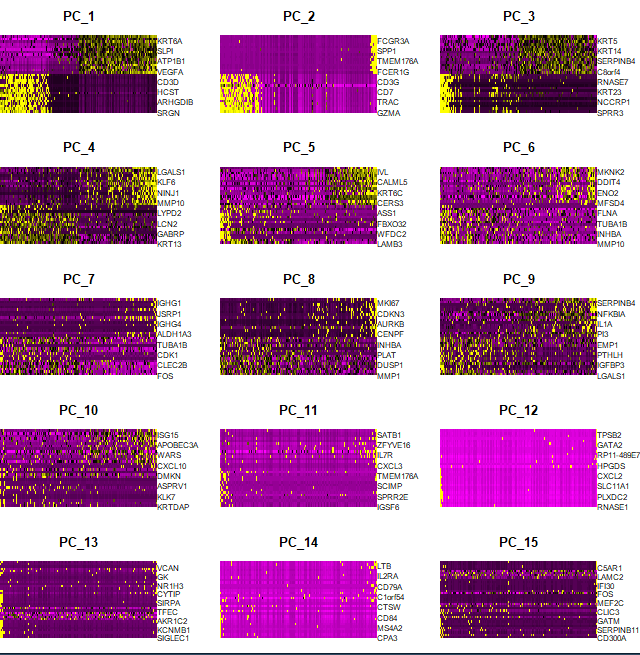
这个就可以看出来一些基因在不同亚群的表达量,但是我们不知道分多少合适呢,这里就需要进行后续的分析:
pbmc<-JackStraw(pbmc,num.replicate = 100)
这一步就是分离分析,小伙伴耐心等待一下
就是在寻找最佳分群
pbmc<-ScoreJackStraw(pbmc,dims=1:20)JackStrawPlot(pbmc,dim=1:20)
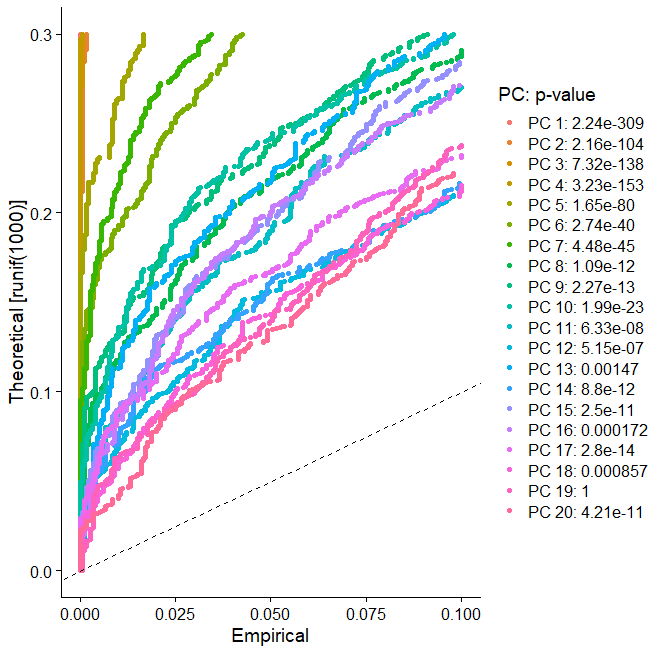
这个就是不同亚群的P值情况,
ElbowPlot(pbmc)
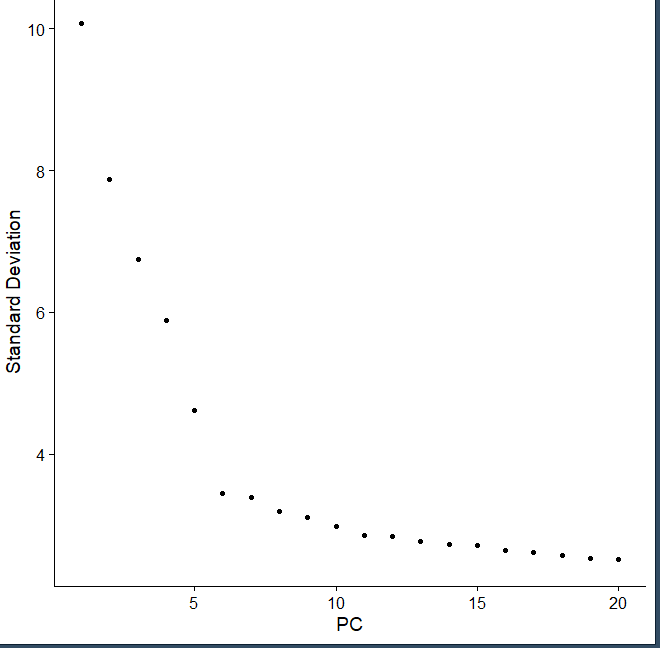
这个碎石图我们就可以看看在哪里平缓我们就选择哪里,小图这张图就选择17或者15都可以
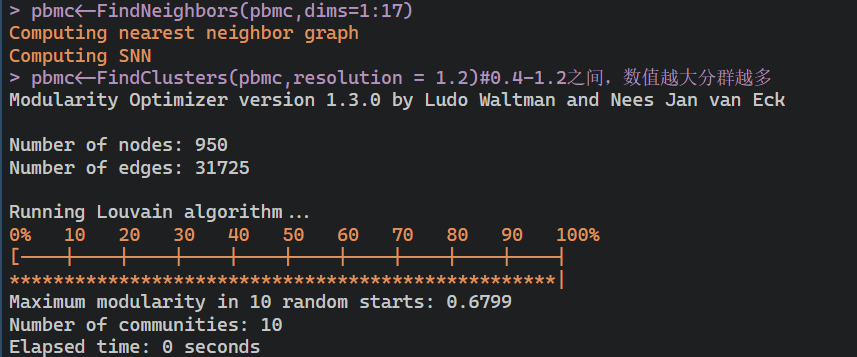
pbmc<-FindNeighbors(pbmc,dims=1:17)pbmc<-FindClusters(pbmc,resolution = 1.2)#0.4-1.2之间,数值越大分群越多
head(Idents(pbmc), 5) 
这里可以看出来我们分了9个亚群
然后我们进行降维
pbmc<-RunUMAP(pbmc,dims=1:17)#一种降维方法
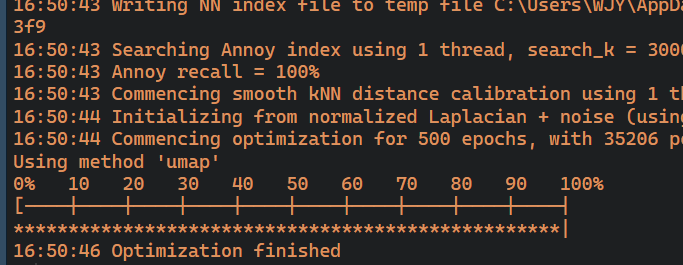
DimPlot(pbmc,reduction = "umap")#umap用于大型数据
然后后面我们就要进行降维绘制图,
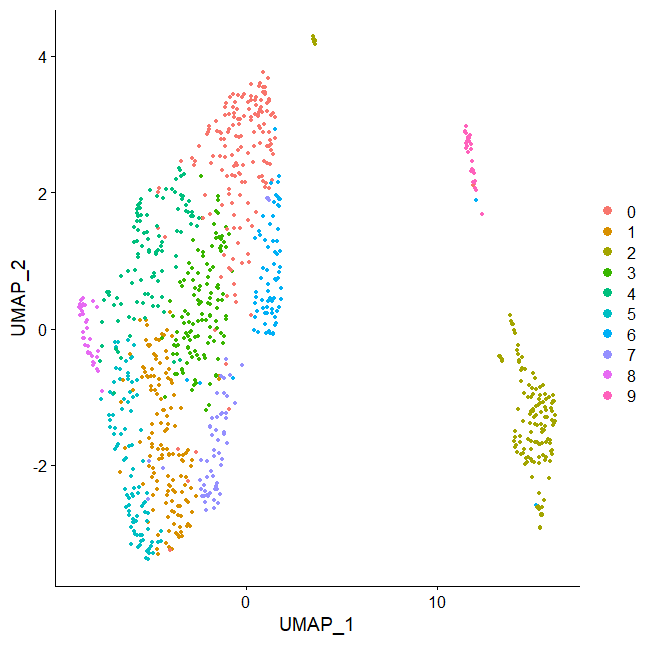
这里用的是umap方式进行可视化降维结果,对于大数据来说是比较适用的,我们还可以进行其他方式,比如tsne,适合小数据
pbmc<-RunTSNE(pbmc,dims=1:17)#另一种降维方法DimPlot(pbmc,reduction = "tsne")#tsne用于小型数据,分离效果更好
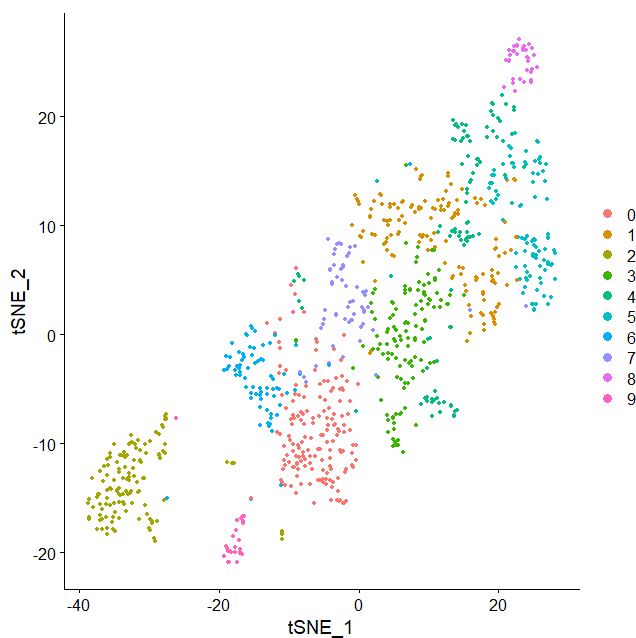
这个就可以看出来,小果的数据是小型数据,所以呢,在使用tsne方式的更明显的分离单细胞亚群
最后呢就是需要找到我们的各类的关键基因
#找各类关键基因pbmc.markers<-FindAllMarkers(pbmc,only.pos = T,min.pct = 0.25,logfc.threshold = 0.25)#寻找所有类的makers基因
pbmc.markers%>%group_by(cluster) %>% top_n(n=2,wt=avg_log2FC)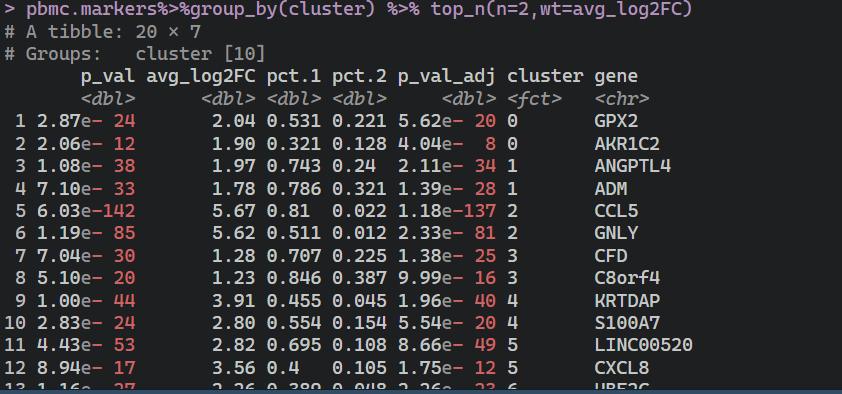
对结果展示,一些关键基因,我们可以选择前十名的展示其在单细胞亚群中的形式
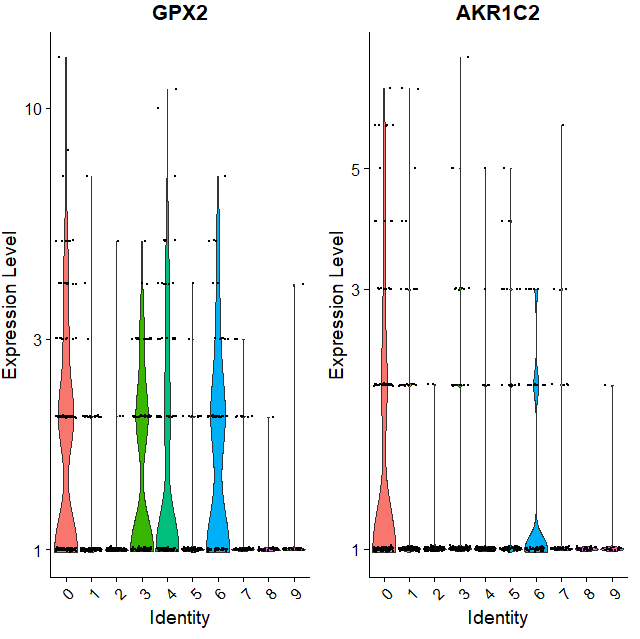
VlnPlot(pbmc,features = c("GPX2","AKR1C2"),slot = "counts",log=T)
小图这里对前两名的基因进行展示
FeaturePlot(pbmc,features = c("GPX2"),reduction = "tsne")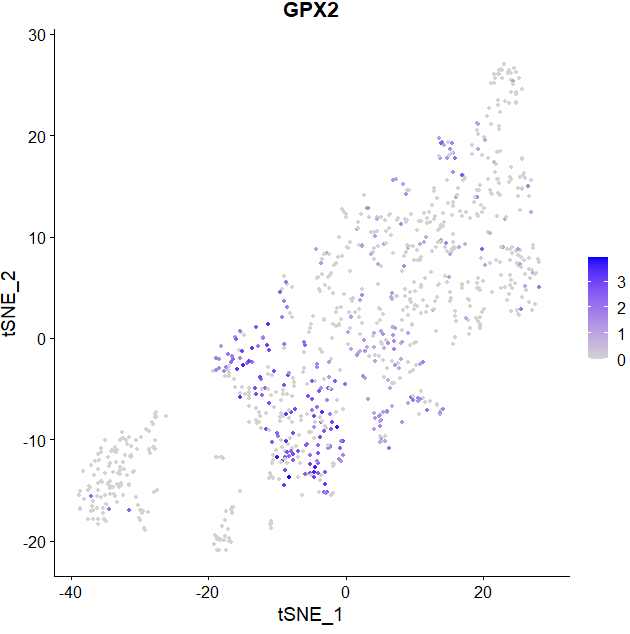
这里就可以很明显的看出GPX2这个基因在那些细胞中占比最多,
reduction = “tsne”这个参数可以改变绘图的方式,
然后我们对前十名的进行保存
top10<-pbmc.markers%>%group_by(cluster) %>% top_n(n=10,wt=avg_log2FC)DoHeatmap(pbmc,features = top10$gene)+NoLegend()
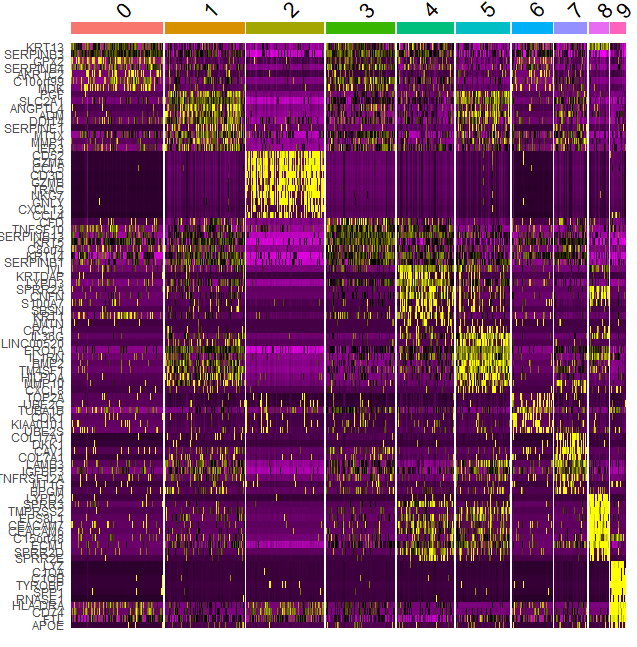
这里对关键基因表达量进行热图的绘制,看看这些基因在那个亚群中表达如何,
saveRDS(pbmc,”pbmc.rds”)
最后我们保存一下这些信息,
然后我们对这些细胞进行注释,这里我们使用SingleR包进行自动注释,比较方便,小果以及把这个数据的信息下载下来了,小伙伴可以自行去后台领取
library(SingleR)BiocManager::install("celldex")library(celldex)library(Seurat)library(pheatmap)
加载一下包,我们进行单细胞注释
##下载注释数据库,这里找小果领取与喜爱下载好的数据库信息
小伙伴也可以自行去下载,不过一般会因为网络问题失败,
hpca.se <- HumanPrimaryCellAtlasData()hpca.se#直接load下载好的数据库
load("HumanPrimaryCellAtlas_hpca.se_human.RData")load("BlueprintEncode_bpe.se_human.RData")
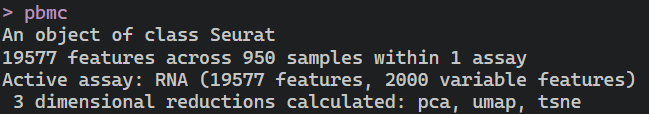
pbmc#上面小果处理好的单细胞信息
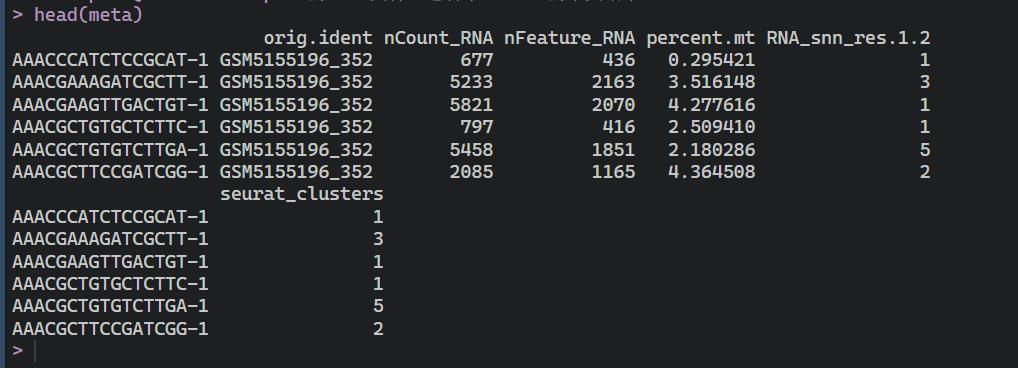
meta=pbmc@meta.data #pbmc的meta文件,包含了seurat的聚类结果head(meta)
这里查看一下信息
plot1 <- DimPlot(pbmc, reduction = "umap", label = TRUE)plot2<-DimPlot(pbmc, reduction = "tsne",label = TRUE)plot1 + plot2
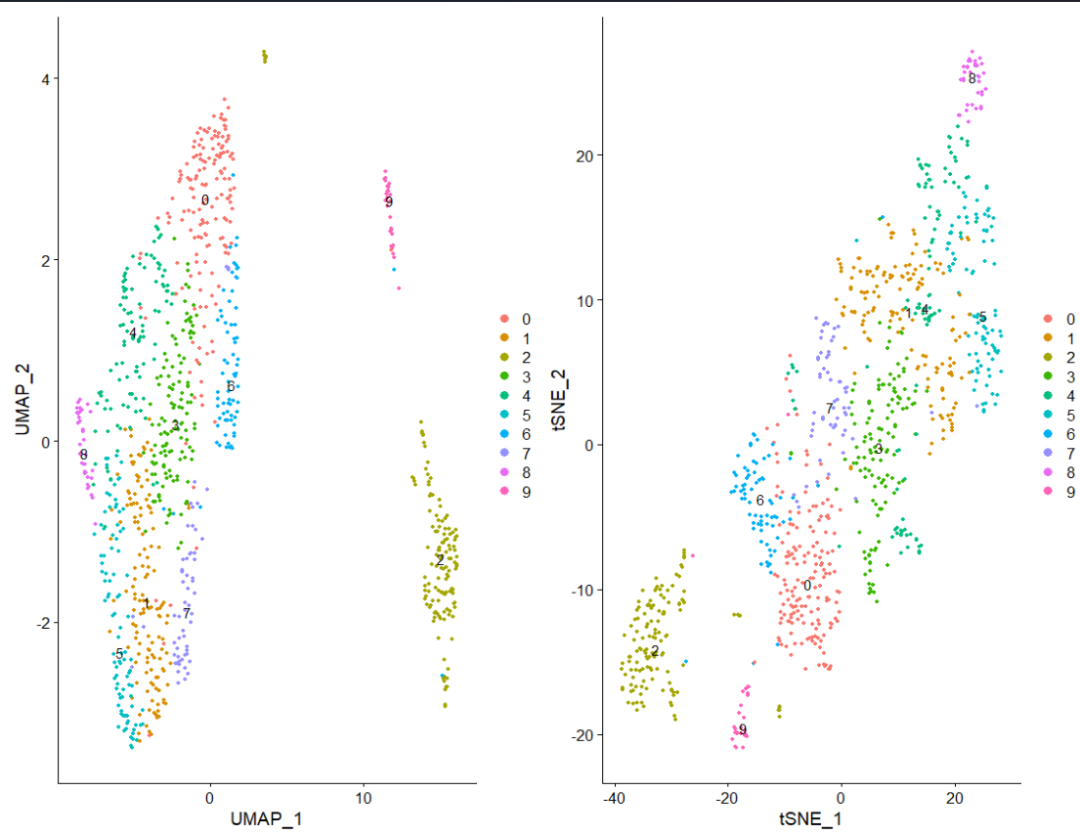
然后我们就开始注释吧!
#进行singleR注释pbmc_for_SingleR <- GetAssayData(pbmc, slot="data") ##获取标准化矩阵pbmc.hesc <- SingleR(test = pbmc_for_SingleR, ref = hpca.se, labels = hpca.se$label.main) #pbmc.hesc
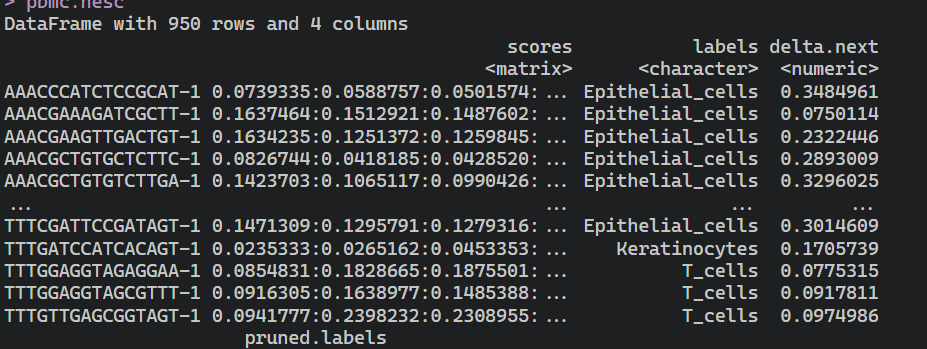
#seurat 和 singleR的table表table(pbmc.hesc$labels,meta$seurat_clusters)
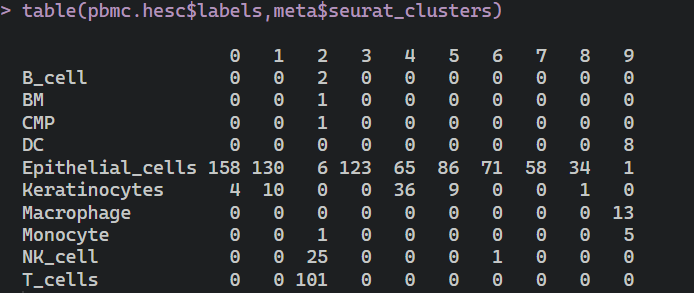
注释结果
#umap图pbmc@meta.data$labels <-pbmc.hesc$labelsprint(DimPlot(pbmc, group.by = c("seurat_clusters", "labels"),reduction = "umap"))
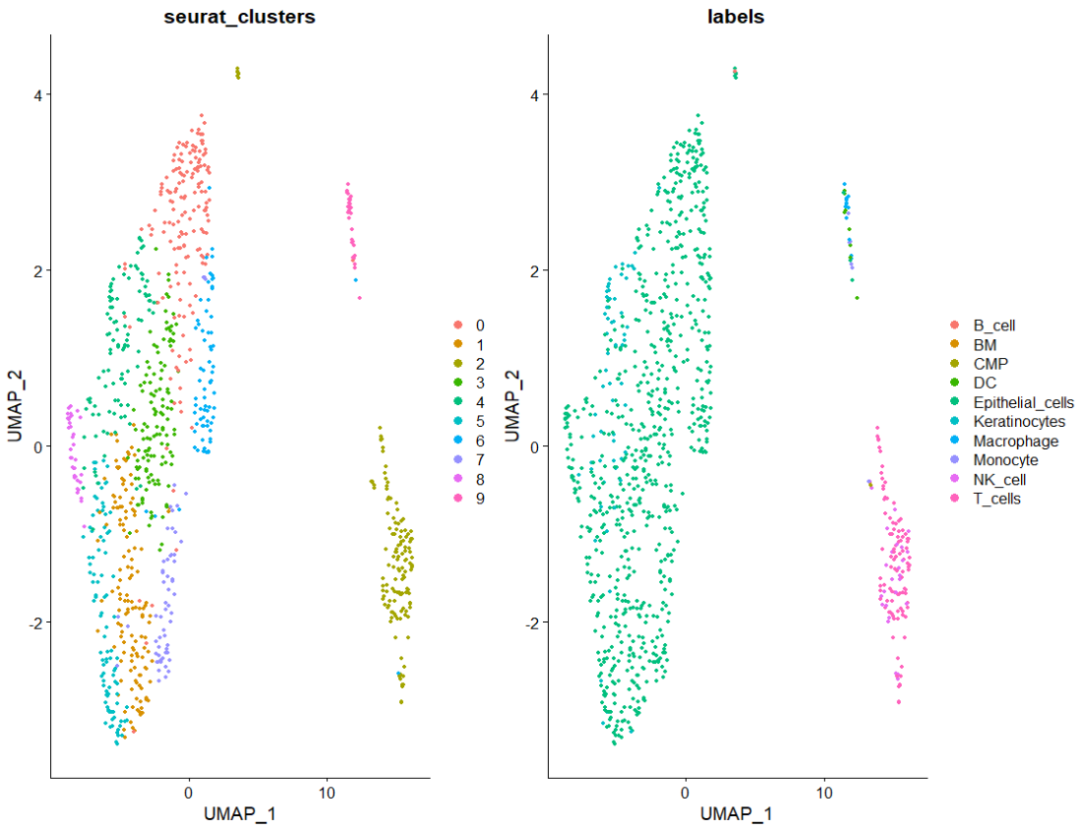
这里我们注释完成了!
但是我们还需要多个数据库验证以及对结果诊断,来看看:
##多个数据库注释pbmc3 <- pbmcpbmc3.hesc <- SingleR(test = pbmc_for_SingleR, ref = list(BP=bpe.se, HPCA=hpca.se),labels = list(bpe.se$label.main, hpca.se$label.main))table(pbmc3.hesc$labels,meta$seurat_clusters)
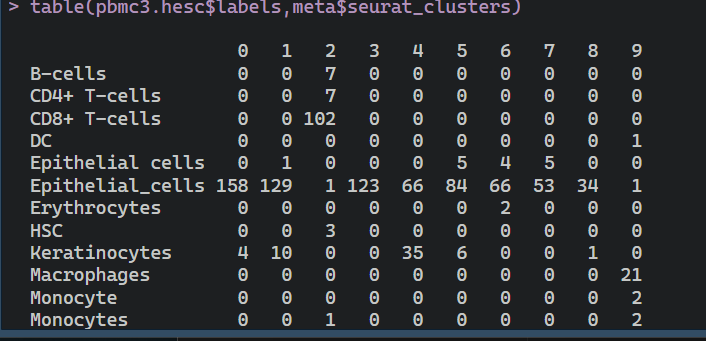
pbmc3@meta.data$labels <-pbmc3.hesc$labels
print(DimPlot(pbmc3, group.by = c("seurat_clusters", "labels"),reduction = "tsne"))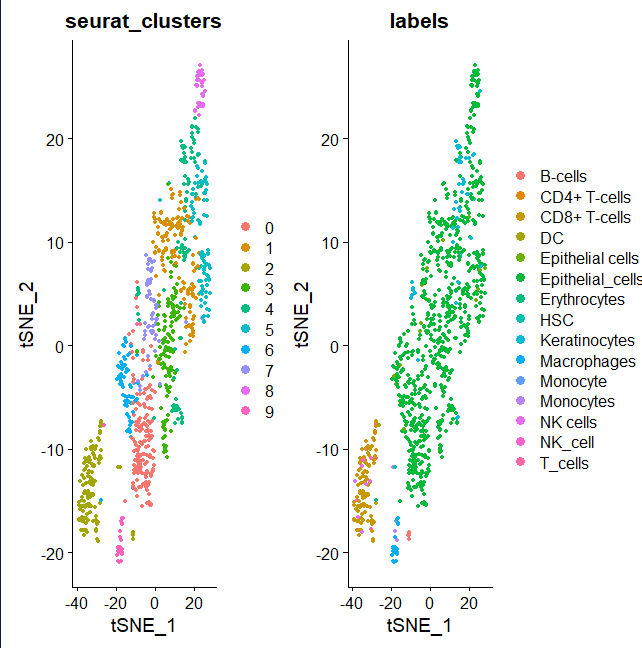
#结果诊断print(plotScoreHeatmap(pbmc.hesc))plotDeltaDistribution(pbmc.hesc, ncol = 3)
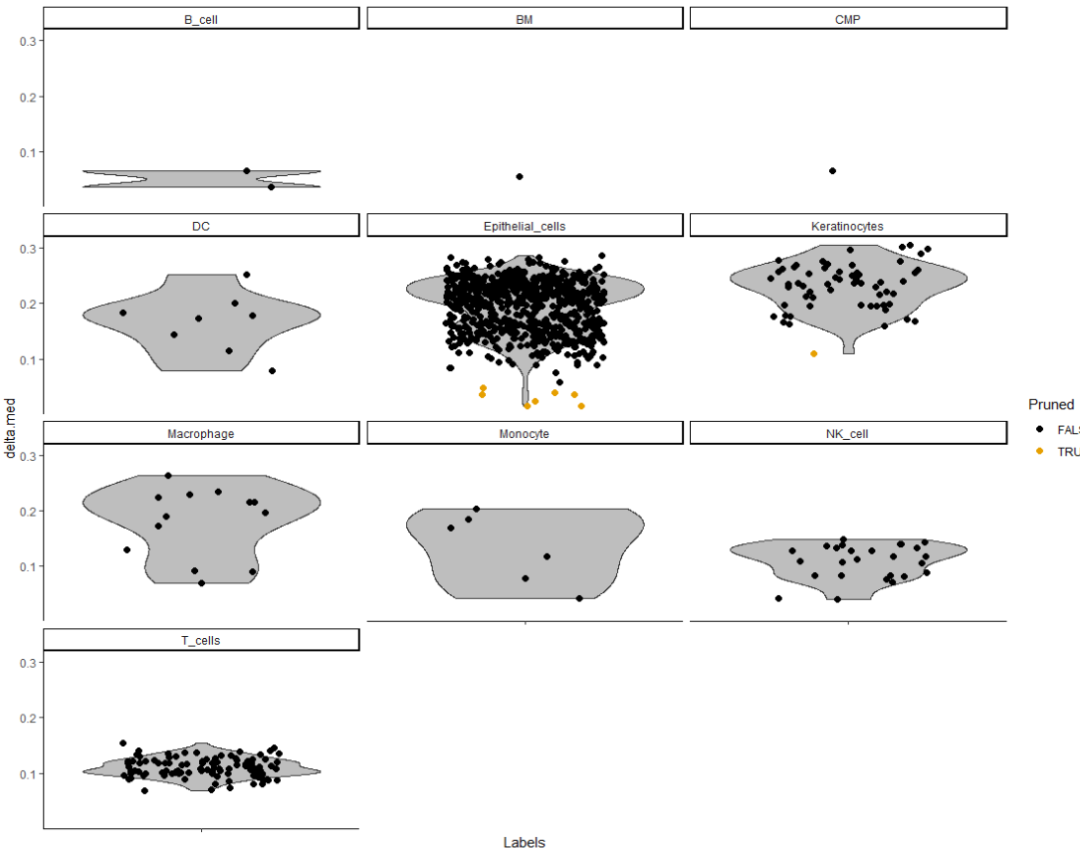
#cluster结果对比tab <- table(label = pbmc.hesc$labels,cluster = meta$seurat_clusters)pheatmap(log10(tab + 10))
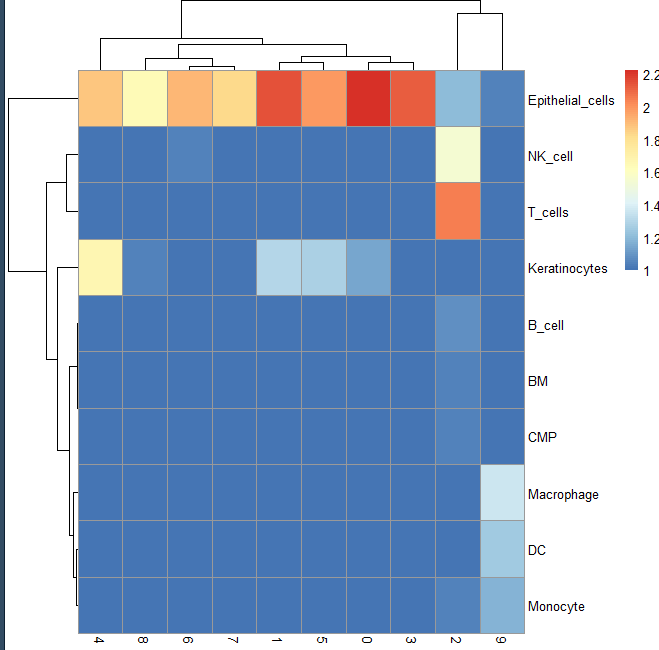
好了,到此我们的单细胞入门学习的教程就结束了,小伙伴要好好消化一下,对此数据进行亲手操作!


往期推荐