纯代码,师妹带你轻松玩转生存分析之独立预后分析
{ 点击蓝字,关注我们 }
之前小师妹带大家做过了森林图的绘制,那么今天我们再来实操生存分析的独立预后分析,寻找与生存期相关的基因,并生成相应的森林图,从而揭示这些基因对生存率的影响。先复习了解一下相关的概念。
独立预后分析: 代码根据每个基因的表达水平,分别构建Cox比例风险模型。这些模型用于评估每个基因是否与生存率相关。如果某个基因的表达与生存率显著相关(满足p值过滤条件),则认为该基因在生存预后中可能具有重要意义。
生存分析: 代码使用survival包对给定的基因表达数据和临床数据进行生存分析。生存分析是一种统计方法,用于评估不同变量(例如基因表达)与生存期之间的关联。通过构建Cox比例风险模型,代码探索了单个基因表达和生存之间的可能关系。
#install.packages('survival')#用library()函数加载survival包。library(survival)expFile="surSigExp.txt"
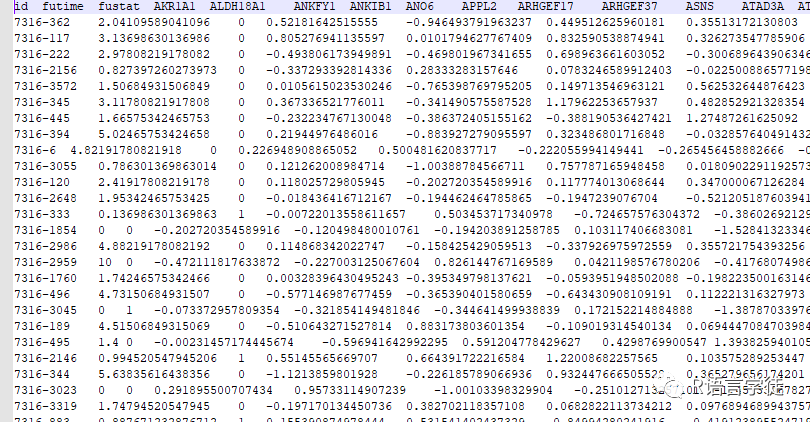
#表达数据文件clinicalFile="clinicalNum.txt"

#临床数据文件#由expFile和clinicalFile变量指定的文件中读取基因表达数据和临床数据。exp=read.table(expFile,sep="t",header=T,check.names=F,row.names=1) #读取表达数据文件cli=read.table(clinicalFile,sep="t",header=T,check.names=F,row.names=1) #读取临床数据文件samSample=intersect(row.names(exp),row.names(cli))exp=exp[samSample,]cli=cli[samSample,]pFilter=0.05 #p值过滤条件#变量pFilter设置为显著性阈值,用于p值############绘制森林图函数############bioForest=function(coxFile=null,forestFile=null,forestCol=null){# 定义自定义函数bioForest,用于从Cox比例风险模型结果创建森林图。# 此函数接受参数,如coxFile(Cox模型结果文件)、forestFile(输出PDF文件)和forestCol(森林图框的颜色)。#读取输入文件rt <- read.table(coxFile,header=T,sep="t",row.names=1,check.names=F)gene <- rownames(rt)#循环遍历表达数据中的每个基因。hr <- sprintf("%.3f",rt$"HR")hrLow <- sprintf("%.3f",rt$"HR.95L")hrHigh <- sprintf("%.3f",rt$"HR.95H")Hazard.ratio <- paste0(hr,"(",hrLow,"-",hrHigh,")")pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))#输出图形pdf(file=forestFile, width = 7,height = 4.5)n <- nrow(rt)nRow <- n+1ylim <- c(1,nRow)layout(matrix(c(1,2),nc=2),width=c(3,2.5))#绘制森林图左边的临床信息xlim = c(0,3)par(mar=c(4,2.5,2,1))plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")text.cex=0.8text(0,n:1,gene,adj=0,cex=text.cex)text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex);text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)text(3,n:1,Hazard.ratio,adj=1,cex=text.cex);text(3,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1,)#绘制森林图par(mar=c(4,1,2,1),mgp=c(2,0.5,0))xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.05,col="darkblue",lwd=2.5)abline(v=1,col="black",lty=2,lwd=2)boxcolor = ifelse(as.numeric(hr) > 1, forestCol, forestCol)points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=1.3)axis(1)dev.off()}############绘制森林图函数#############独立预后分析,输出表格allOutTab=data.frame()#初始化一个空的数据框 allOutTab,用于存储独立预后分析的结果sigGenes=c("futime","fustat")#一个字符向量 sigGenes,其中包含初始的两个列名 "futime" 和 "fustat",这些列用于构建 Cox 比例风险模型。for(i in colnames(exp[,3:ncol(exp)])){#循环遍历表达数据的列,其中包括具体的基因表达信息。#针对每个基因,构建 Cox 比例风险模型,并对模型的结果进行总结(summary)。rt=cbind(exp[,1:2],cli,gene=exp[,i])colnames(rt)[ncol(rt)]=icox <- coxph(Surv(futime, fustat) ~ rt[,ncol(rt)], data = rt)coxSummary = summary(cox)outTab=data.frame()# p 值小于预设的过滤条件(pFilter),则继续在内部循环中,分别构建只包含一个基因表达变量的 Cox 比例风险模型。if(coxSummary$coefficients[,"Pr(>|z|)"]for(j in colnames(rt[,3:ncol(rt)])){coxJ <- coxph(Surv(futime, fustat) ~ rt[,j], data = rt)coxSummaryJ = summary(coxJ)coxP=coxSummaryJ$coefficients[,"Pr(>|z|)"]if(coxSummaryJ$conf.int[,"upper .95"] != Inf){outTab=rbind(outTab,cbind(id=j,HR=coxSummaryJ$conf.int[,"exp(coef)"],HR.95L=coxSummaryJ$conf.int[,"lower .95"],HR.95H=coxSummaryJ$conf.int[,"upper .95"],pvalue=coxSummaryJ$coefficients[,"Pr(>|z|)"]))}}write.table(outTab,file="uniCox.txt",sep="t",row.names=F,quote=F)bioForest(coxFile="uniCox.txt",forestFile=paste0("uni.",i,".pdf"),forestCol="green")unlink("uniCox.txt")sigGenes=c(sigGenes,i)allOutTab=rbind(allOutTab,cbind(id=i,HR=coxSummary$conf.int[,"exp(coef)"],HR.95L=coxSummary$conf.int[,"lower .95"],HR.95H=coxSummary$conf.int[,"upper .95"],pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))}}#输出单因素独立预后分析结果write.table(allOutTab,file="uniIndep.xls",sep="t",row.names=F,quote=F)#输出单因素独立预后基因表达量indepSigExp=exp[,sigGenes]indepSigExp=cbind(id=row.names(indepSigExp),indepSigExp)write.table(indepSigExp,file="uniIndepSigExp.txt",sep="t",row.names=F,quote=F)

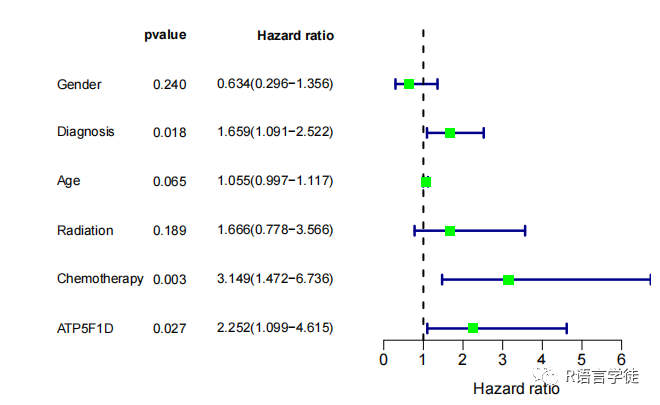

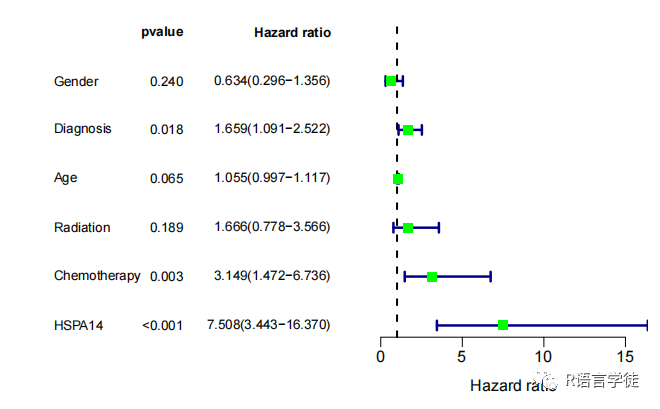
将独立预后分析的结果写入名为 “uniIndep.xls” 的文件中,其中包括每个基因的Hazard Ratio、置信区间和p值等信息。此外,代码还将满足显著性条件的基因的表达数据写入名为 “uniIndepSigExp.txt” 的文件中。
通过生存分析和独立预后分析,可以识别出在生存率方面具有显著影响的基因。这些基因可能在疾病进程、治疗反应等方面起着重要作用。可以进一步分析这些基因的功能、通路以及与疾病相关的机制,下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:
http://www.biocloudservice.com/home.html。
云生信平台链接:
http://www.biocloudservice.com/home.html。

END

