如此简单吗?一文拿捏GEO数据库芯片数据下载及差异分析
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

通过该推文,你将完全掌握GEO公共数据下载和差异分析,非常非常适合小白,如果你有数据挖掘的想法,接下来马上跟着小果开始今天的学习之旅,相信你会收获满满。
1. 如何获得相关疾病的GEO数据库ID
在进行实操之前,最重要的是要获得相关疾病的GEO数据库ID,如何获得呢?不慌!小果为大家介绍两种常用的方法。第一种方法是通过NCBI GEO DataSets 数据库下载,可以直接输入想查询的疾病名称就可以进行搜索获得相应的数据,网址为:https://www.ncbi.nlm.nih.gov/gds/?term=GSE70494

第二种方法是通过以发表的文献来查询相关疾病的GEO ID,小果是通过PubMed来查询,只需要输入相关疾病关键字和GEO就可以搜索到相关文章和GEO ID,该网址为:https://pubmed.ncbi.nlm.nih.gov/?term=gene+family
一般通过这两种方法就可以获取自己想要的GEO ID,通过小果的方法获得ID后,就可以跟着小果开始今天的实操分析啦,其实就是这么简单!!!哈哈哈哈哈。。。。。。。。
2. 导入需要的R包
library(GEOquery)library(limma)
3. 从GEO下载表达矩阵和芯片注释注释文件
GSEdata <- getGEO("GSE17215", GSEMatrix=T, AnnotGPL=FALSE)eset <- GSEdata[[1]]#提取表达矩阵,通过exprs函数提取,行名为探针ID,列名为样本名exprSet <- as.data.frame(exprs(eset))

# 提取GSM ID作为样品名,pData函数来获取样本信息GSMID <- pData(eset)[,c(1,2,8,10,12)]
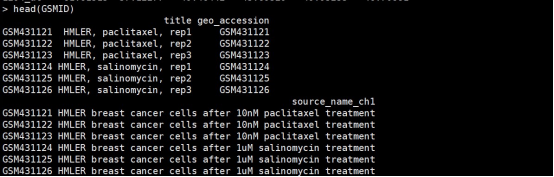
#整理芯片注释信息colnames(exprSet) <- GSMID$geo_accession# 获得探針组跟gene symbol的对应关系gpl <- getGEO(eset$platform_id[1])dim(Table(gpl)) #查看一共有多少行探针组# 提取自己想要的信息,此处第1、11、12列,分别是行名ID和其对应的Gene Symbol和ENTREZ_GENE_IDannotation <- Table(gpl)[,c(1,11,12)]

#将factor 改为 characteri <- sapply(annotation, is.factor) # Change factor to characterannotation[i] <- lapply(annotation[i], as.character)# 有些探针组对应多個基因,使用“///”分割,當然你可以选择保留,我的习惯是保留第一个annotation[,2]<-data.frame(sapply(annotation[,2],function(x) unlist(strsplit(x,'///'))[1]),stringsAsFactors = F)[,1]annotation[,3]<-data.frame(sapply(annotation[,3],function(x) unlist(strsplit(x,'///'))[1]),stringsAsFactors = F)[,1]# 把整理好的探针组ID、gene symbol、ENTREZ ID对应关系保存到文件里colnames(annotation) <- c("probe_id", "gsym", "entrez_id")write.csv(annotation, "gplTOgene.csv", row.names = F)
#小果来画重点啦!不同的测序平台芯片注释信息可能有差异奥,在提取注释信息的时候要根据不同平台格式灵活处理,本实例数据测序平台为GPL3912,可以点击查看注释信息内容。点击GPL3912,就可以看到芯片完整注释信息:


4.把表达矩阵的探针ID换成Gene Symbol
annotation <- read.csv("gplTOgene.csv")head(annotation)

#将探针信息和表达矩阵合并exprSet$probe_id <- rownames(exprSet)express <- merge(x = exprSet, y = annotation[,c(1:2)], by = "probe_id", all.x = T)

#去除probe_id这列信息express$probe_id <- NULL# 有些gene对应多个探针组,此处保留其中表达量最大值rowMeans <- apply(express, 1, function(x) mean(as.numeric(x), na.rm = T))express <- express[order(rowMeans, decreasing =T),]express <- express[!duplicated(express[, dim(express)[2]]),]express <- na.omit(express)dim(express)
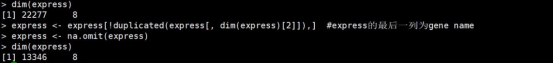
#将最后一列作为行名rownames(express) <- express[,dim(express)[2]]#删除最后一列express <- express[,-(dim(express)[2])]# 表达矩阵(express)已构建好,保存到文件write.csv(express, "easy_input_expr.csv", quote = F)
5.按照实验设计整理分组信息
pdata <- pData(eset)i <- sapply(pdata, is.factor)pdata[i] <- lapply(pdata[i], as.character)# 按照实验设计,构建分组矩阵group_list <- subset(pdata, select = title) # Sample的分组信息group_list$condition = rep(c("a","b"), each = 3)
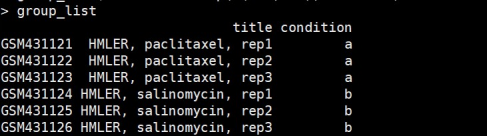
#分组矩阵(design)已构建好
write.table(group_list, "easy_input_pheno.txt", quote = F, sep = "t", row.names = F)6.利用limma 包进行差异分析
#不做均一化处理express.norm <- express#生成模型设计矩阵design <- model.matrix(~ 0 + factor(group_list$condition))#替换列名colnames(design) <- levels(factor(group_list$condition))#替换行名rownames(design) <- colnames(express.norm)

#构建比较矩阵contrast.matrix <- makeContrasts(b-a, levels = design)

#线性模型拟合fit <- lmFit(express.norm,design)#计算estimated coefficient 和标准误差fit2 <- contrasts.fit(fit,contrast.matrix)#差异分析fit2 <- eBayes(fit2)#提取所有差异基因表格,行名为Gene symbol,列名主要关注logFC,P.Value和adj.P.Val,通过pvalue值排序。x <- topTable(fit2, coef = 1, n = Inf, adjust.method = "fdr", sort.by = "P")
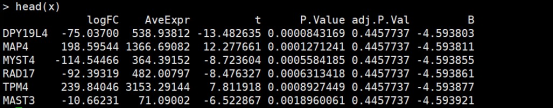
#把全部基因的limma分析结果保存到文件
write.csv(x, "limma.csv", quote = F)7.结果文件
1. gplTOgene.csv
该结果文件为探针id对应的Gene symbol和entrez_id,第一列为探针ID,第二列为Gene symbol,第三列为entrez_id。

2. easy_input_expr.csv
该结果文件为处理好的表达矩阵文件,行名为Gene symbol,列名为对应的样本名。
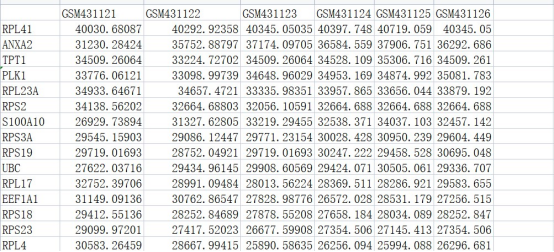
3. easy_input_pheno.txt
该结果文件为提取的样本分组文件。

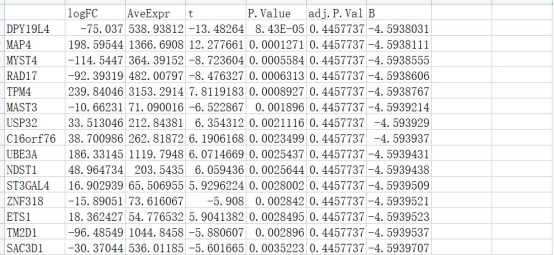
4. easy_input_limma.csv
该结果文件为通过limma包进行差异分析结果文件,行名为Gene symbol,第一列为log2FC,第四列为Pvalue值,第五列为矫正后的Pvalue值。
今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:http://www.biocloudservice.com/home.html,包括了GEO数据下载(http://www.biocloudservice.com/371/371.php),limma+gsva(http://www.biocloudservice.com/371/371.php)等小工具,欢迎大家和小果一起讨论学习哈!!!!

扫码加小果
领取生信大礼包
点击“阅读原文”立刻拥有
↓↓↓