不可多得的GISTIC2.0教学之TCGA拷贝数变异CNV数据整理
点击蓝字 关注我们
小伙伴们大家好,我是大海哥,今天大海哥发现最近很多文章都结合了拷贝数变异来对癌症机制进行解释,但是我百度一查发现了一些问题,很多小伙伴都不知道该如何去对拷贝数变异数据展开分析,甚至都不知道该从哪里下载对应的数据,基于此类现象,大海哥决定出一个关于分析拷贝数变异的详细教程,同时使用在线分析工具GISTIC2.0来展开分析,希望可以帮到大家!
首先,我们来了解一下拷贝数变异是什么?
拷贝数变异(Copy Number Variation,CNV)
是指一个基因组中特定基因或基因组区域的拷贝数(复制数)在不同个体之间发生变化的现象。正常情况下,人类每个细胞都应该包含一份完整的基因组,即每个基因或基因组区域的拷贝数为2(两个等位基因,一个来自母亲,一个来自父亲)。然而,由于基因组复制和维护的不稳定性,有时就会发生拷贝数的变化。并且它们可能对个体的表型特征、疾病易感性以及药物反应等产生影响
了解了CNV是什么,那我们就开始来了解一下如何处理CNV数据吧。
首先我们要从TCGA中下载对应的CNV数据,一共有两种,分别为Copy Number Segment和Masked Copy Number Segment,我们看看官方解释:
Copy Number Segment:将连续染色体片段与基因组坐标、平均阵列强度以及与每个片段结合的探针数量相关联的表格。
Masked Copy Number Segment:删除了带有已知含有种系突变的探针的片段,且与拷贝数片段具有相同信息的表
然后我们本次分析选择Masked Copy Number Segment 数据,是一个gz格式的压缩包,我们解压后进行处理。
上代码!
rm(list = ls())## 将整理好的Masked Copy Number Segment重新命名为“rawdata”length(dir("rawdata") )#创建一个用来放txt文件的文件夹,因为解压后还有多个压缩包,每个压缩包包含一个txt文件dir.create("rawdata_all")#用lapply将rawdata里面txt文件整理到一个文件夹里raw_txt <- dir("rawdata/")lapply(raw_txt, function(x){mydir <- paste0("./rawdata/",x)file <- list.files(mydir,pattern = "nocnv_grch38")myfile <- paste0("./rawdata/",x,"/",file)file.copy(myfile,"rawdata_all")})
最后会得到多个txt文件,如下图
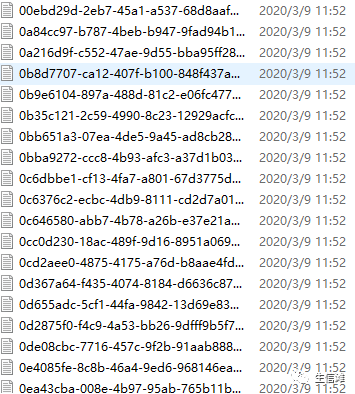
#然后开始整合啦!library(data.table)cnv_file <- dir("rawdata_all")cnv_file[1]
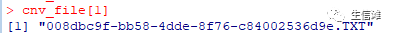
file <- paste0("./rawdata_all/",cnv_file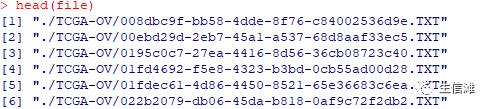
## 批量读取txt文件file_list <- list()fred_cnv <- function(x){file_list[[x]] <- data.table::fread(file = x,data.table = F)file_list[[x]]}library(dplyr)cnv_df <- lapply(file, fred_cnv)#行合并cnv_df <- do.call(bind_row,cnv_df)head(cnv_df)
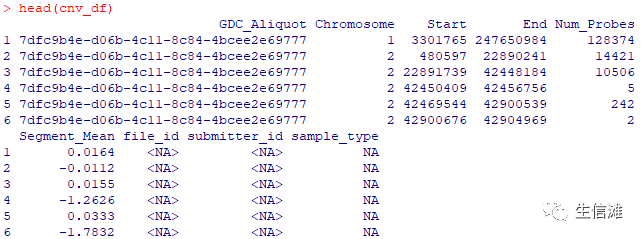
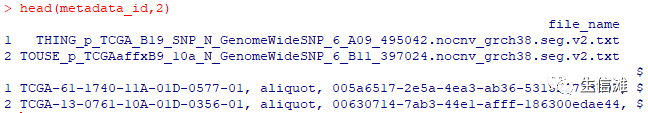
## 读取metadata里面的注释信息metadata <- jsonlite::fromJSON("metadata.cart.2021-02-15.json")metadata_id <- metadata %>%dplyr::select(c(file_name,associated_entities))meta_df <- do.call(rbind,metadata$associated_entities)head(metadata_id,2)
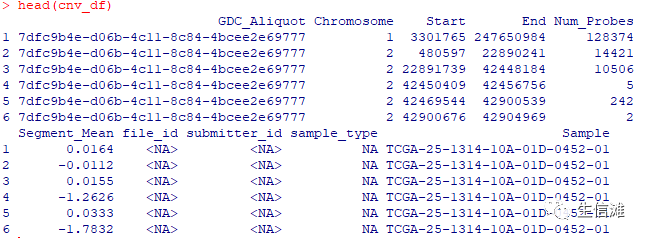
cnv_df$Sample <- meta_df$entity_submitter_id[match(cnv_df$GDC_Aliquot,meta_df$entity_id)]length(unique(cnv_df$GDC_Aliquot))## [1] 1025length(unique(cnv_df$Sample))## [1] 1025head(cnv_df)
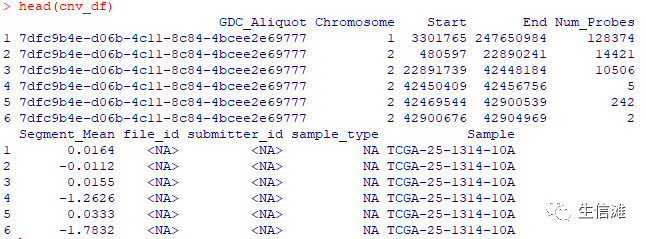
#把样本名改一下cnv_df$Sample <- substring(cnv_df$Sample,1,16)head(cnv_df)
## Gistic2.0只需要maskedCNS的六列cnv_df <- cnv_df[,c('Sample','Chromosome','Start','End','Num_Probes','Segment_Mean')]head(cnv_df)

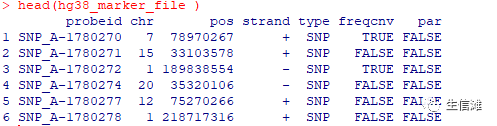
#好了,整理完毕,现在这个数据就可以直接拿去分析啦!#别着急!还有一个数据需要整理呢!因为GISTIC2.0需要两个输入文件,我们之整理了一个,还有一个marker file数据需要整理#需要在https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files中下载最新版本的“SNP6 GRCh38 Remapped Probeset File for Copy Number Variation Analysis”文件,名字为snp6.na35.remap.hg38.subset.txt.gz。#下载后,开始代码处理部分吧hg38_marker_file <- read.delim("snp6.na35.remap.hg38.subset.txt.gz")head(hg38_marker_file )
#根据官方提示If you are using Masked Copy Number Segment for GISTIC analysis, please only keep probesets with freqcnv = FALSE#我们只用保留freqcnv = FALSE的数据就好啦!hg_marker_file <- hg38_marker_file[hg38_marker_file$freqcnv=="FALSE",]hg_marker_file <- hg_marker_file [,c(1,2,3)]
然后就好啦,只用这三列就可以了。
现在我们就获得了两个数据,一个CNV数据和一个marker file数据,只需这两个文件,我们就可以使用GISTIC2.0展开分析啦。


点击“阅读原文”进入网址