三行代码解决聚类问题!层次聚类你心动了吗
{ 点击蓝字,关注我们 }
层次聚类在生物信息学中广泛应用于挖掘基因表达谱、蛋白质组学、代谢组学等高通量数据的模式识别和数据挖掘。具体而言,层次聚类可以将大量的高维数据转化为低维的聚类树,从而揭示数据之间的内在关系和相似性。
在基因表达谱聚类中,层次聚类可以将基因按照其表达模式分成不同的簇,从而发现具有类似表达模式的基因集合,进一步挖掘基因调控网络和生物学意义。
在蛋白质组学聚类中,层次聚类可以将蛋白质按照其功能类别和表达模式分成不同簇,进一步研究蛋白质功能和相互作用网络。在代谢组学聚类中,层次聚类可以将代谢产物按照其代谢途径和生理功能分成不同簇,进一步研究代谢网络和生物学意义。在生物序列聚类中,层次聚类可以将不同序列按照其相似性和功能特征分成不同簇,进一步研究序列演化和生物学意义。
层次聚类结果的可视化通常是一棵树形结构,也称为树状图(dendrogram)。在树状图中,每个节点代表一个聚类簇,节点的高度表示该聚类簇与其它聚类簇的距离或相似性。树状图的根节点代表所有样本的聚类簇,叶节点代表单个样本。

树状图的横轴通常表示样本或特征,纵轴表示距离或相似性。在树状图中,每个样本或特征在叶节点处,其所属的聚类簇可以通过根节点到叶节点的路径来确定。在树状图中,聚类簇的数量由横轴上的划分线(或虚线)决定,可以通过调整划分线的位置来控制聚类簇的数量。
在观察树状图时,可以通过以下方式来解读聚类结果:
-
根据聚类簇的高度来确定聚类簇的相似性。高度越低,表示聚类簇之间的相似性越高,反之亦然。
-
根据划分线的位置来确定聚类的数量。划分线越高,表示聚类簇数量越少,反之亦然。
-
根据样本或特征在树状图中的位置来确定其所属的聚类簇。同一个聚类簇内的样本或特征位于同一子树中。
-
可以通过颜色或标记来区分不同的聚类簇。在树状图中,同一聚类簇内的样本或特征通常使用相同的颜色或标记。
总之,在观察树状图时,需要结合聚类算法和距离度量方法来理解聚类结果。同时,需要根据具体的研究问题和数据特征来选择适当的聚类算法和距离度量方法,以获得更可靠和准确的聚类结果。
现在小师妹就用R语言手把手带你使用层次聚类
分析~
“hclust”函数是R语言中的一个层次聚类函数,用于对数据进行层次聚类分析。该函数基于距离矩阵,可以通过不同的距离度量方法(如欧氏距离、曼哈顿距离、皮尔逊相关系数等)来计算样本之间的距离。在进行层次聚类时,该函数可以使用不同的聚类算法(如单链接、完全链接、平均链接等)来计算样本之间的相似性,从而将样本分成不同的簇。
“hclust”函数的基本语法如下:
hclust(d, method = "complete")其中,d是一个距离矩阵或相似性矩阵,method是聚类算法的选择(默认为“complete”,即完全链接算法)。该函数的返回值是一个树形结构,表示样本之间的层次聚类关系。可以通过plot函数将树形结构可视化,从而更好地理解聚类结果。
除了基本语法外,”hclust”函数还可以通过其他参数进行定制化设置,如:
“method”参数可以设置聚类算法的选择,包括单链接算法(”single”)、完全链接算法(”complete”)、平均链接算法(”average”)等;
“members”参数可以设置每个簇的最小样本数;
“dist.method”参数可以设置距离度量方法的选择,包括欧氏距离(”euclidean”)、曼哈顿距离(”manhattan”)等。
“hclust”函数在生物信息学中广泛应用于基因表达谱、蛋白质组学、代谢组学等高通量数据的聚类分析。通过”hclust”函数,可以将样本或特征分成不同的簇,揭示数据之间的内在关系和相似性,进一步挖掘数据的生物学意义。
以biopsy数据为例,首先我们对数据进行预处理,将变量的类型转换成数值类型,提取出只含有特征变量的矩阵。
mat <- as.matrix(biopsy)duplicated_rows <- duplicated(mat[,2:10])duplicate_indices <- which(duplicated_rows)mat <- mat[-duplicate_indices, ]mat <- mat[complete.cases(mat), ]mat1=mat[,2:10]mat1<-apply(mat1, 2, as.numeric)
接下来呢,我们就可以使用欧几里得距离度量方法来计算样本间距离
dist_matrix <- dist(mat1, method = "euclidean")这里我们使用Ward方法进行层次聚类
hc <- hclust(dist_matrix, method = "ward.D2")现在我们就可以可视化我们的聚类结果
plot(hc, labels=FALSE, axes=FALSE)
还可以对聚类结果画热图
# 将层次聚类结果划分为3个聚类簇clusters <- cutree(hc, k = 3)# 将聚类簇标签与原始数据矩阵合并mat_clusters <- cbind(mat1, clusters)# 画热图heatmap(mat_clusters, scale = "row", Rowv = FALSE, Colv = FALSE, col = c("red","green","blue"))
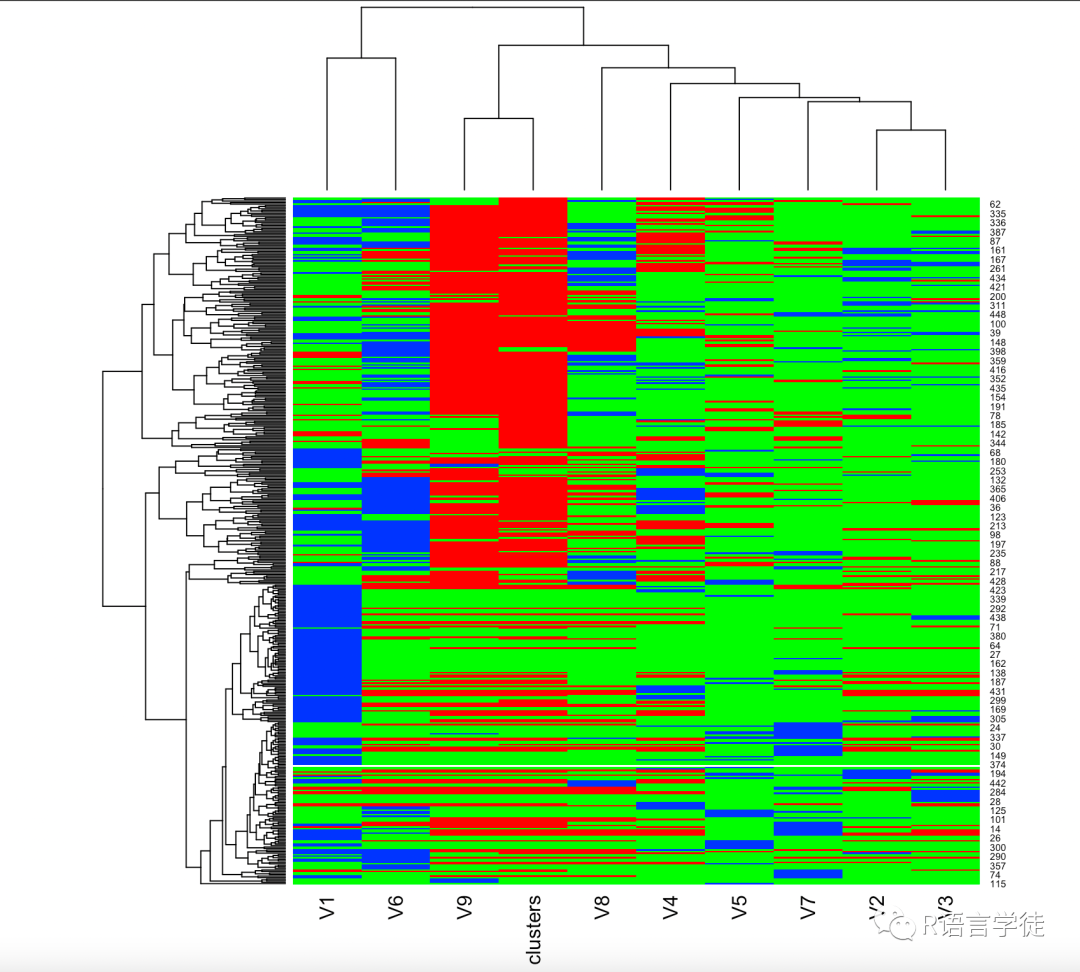
其中每个样本对应一个行,每个聚类簇对应一种颜色,可以直观地展示样本之间的相似性和聚类簇之间的差异。
小师妹今天主要介绍了层次聚类(Hierarchical Clustering)算法的基本原理、应用场景和实现方法。层次聚类是一种无需预先指定聚类簇数量的聚类方法,可以根据数据的内在结构和相似性将样本或特征分成不同的簇,通常用于数据挖掘、生物信息学、社会网络分析等领域。
更多方便实用的小工具在云生信平台等着你哦
http://www.biocloudservice.com/home.html
E
N
D
