三分钟速通featureCounts包——获得 raw counts 无缝衔接差异表达分析
点击蓝字 关注我们
转录组上游分析中,我们需要对reads进行精确的计数,通过获得原始计数数据,我们可以进行后续的差异表达分析、基因表达量比较等定量分析。今天小师妹给大家介绍的featureCounts工具就可以轻松实现这个功能。
这个工具是什么?
featureCounts是一种常用的基因计数工具,用于从高通量测序数据中获得基因或转录本的计数信息。它可以处理RNA-seq或ChIP-seq等测序数据,并将reads根据其在基因或转录本上的映射位置进行计数。该软件的主要功能是将每个基因或转录本与reads关联,并生成计数矩阵,其中每行表示一个基因或转录本,每列表示一个样本,单元格中的值表示该基因或转录本在相应样本中的reads数。
featuresCounts通常与测序数据比对软件(例如STAR、HISAT2等)结合使用,这些软件用于将reads映射到基因组或转录组上。一旦reads被比对上,featuresCounts会根据比对结果进行计数,从而得到每个基因或转录本的reads数。
接下来就跟着小师妹一起实践起来吧!
下载安装
软件官网:The Subread package (sourceforge.net)
可以直接使用以下命令进行下载
wget https://jaist.dl.sourceforge.net/project/subread/subread-1.6.0/subread-1.6.0-Linux-x86_64.tar.gztar -zxvf subread-1.6.0-Linux-x86_64.tar.gz
输入输出数据
要使用featureCounts软件,我们需要提供以下输入数据:
a:读段映射文件(Alignment file):这是由测序数据比对软件生成的文件,通常是BAM格式或SAM格式的文件。该文件包含了测序数据中每个read的映射位置信息。
b:注释文件(Annotation file):这是一个描述基因或转录本位置和特征的文件,通常是GTF(基因组注释文件)或SAF(简化注释格式)格式的文件。注释文件包含了基因或转录本的坐标信息,用于将读取与相应的基因或转录本关联起来。
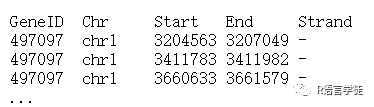
图1 SAF文件格式(列以制表符分隔)
除了这些必需的输入数据之外,您还可以提供一些可选的参数,以根据您的分析需求进行自定义。这些参数可以控制计数的特定设置,如是否考虑多个映射位置、是否考虑链特异性等。
参数介绍
featureCounts软件提供了多个参数,用于控制计数过程和定量分析的各个方面。以下是featureCounts的一些常用参数的简要介绍:
-a
-o
-t
-g
-s
-p:用于开启多线程处理,加快计数的速度。您可以指定使用的线程数。
-M:用于考虑多个映射位置。默认情况下,featureCounts只计数唯一映射的读取,使用该参数可以启用多个映射位置的计数。
-B:用于计算基于区间的计数。这个参数可以用于处理重叠的注释区域,确保一个读取只被计数一次。
当然这只是featureCounts可用参数的一小部分,还有其他参数可以用于更详细的设置和分析,小师妹建议您可以参考featureCounts的官方文档或帮助文档。更多学习资源请大家移步小师妹专属云生信平台(云生信 – 学生物信息学 (biocloudservice.com))搜索更多资源哦!
E
N
D

师妹微信
扫码添加