两行代码10+张图的聚类神器ConsensusClusterPlus(上)
{ 点击蓝字,关注我们 }
在生物信息分析的过程中,我们常常需要用到聚类分析,今天小师妹就给大家带来一个聚类神器ConsensusClusterPlus~
ConsensusClusterPlus是一个基于共识聚类(Consensus Clustering)的R包,用于对高维数据进行聚类分析。共识聚类是一种基于重复抽样的聚类方法,它通过对多次随机抽样得到的数据子集进行聚类分析,并将多次聚类结果进行整合,从而得到更加鲁棒和稳定的聚类结果。
ConsensusClusterPlus包拥有多种聚类算法选项,包括k-means、hierarchical clustering、PAM等。该包支持在不同聚类数(k值)下运行共识聚类,并且能够自动选择最优的聚类数,同时还提供了可视化工具,例如跟踪图、共识矩阵等,方便用户对聚类结果进行分析和解释。
在使用ConsensusClusterPlus时,需要设置一些参数以获得最佳的聚类结果。下面是ConsensusClusterPlus中常用的参数介绍:
d:要聚类的数据;要么是一个数据矩阵,其中 columns=items/samples 和 rows 是特征。例如,行中基因和列中微阵列的基因表达矩阵,或 ExpressionSet 对象,或距离对象(仅适用于没有特征重采样的情况)
distance:距离度量方法,可以是欧几里得距离(”euclidean”)、相关系数(”correlation”)、皮尔逊相关系数(”pearson”)等。
maxK:聚类数范围,最大聚类数,需要是一个整数值。
seed:为可重现的结果设置随机种子。
reps:重复次数,表示在每个聚类数下进行的随机抽样次数。
pItem:项目(列)的子集大小,表示在每个重复中要抽样的特征数,通常设置为数据集的平方根。
clusterAlg:聚类算法,可以是k-means(”km”)、hierarchical(”hc”)、PAM(”pam”)等。
pItemReplace:在每个重复中是否替换选定的项目(列)。
verbose:是否显示迭代过程的详细信息,如果为 TRUE,则将消息打印到屏幕以指示进度。这对于大型数据集很有用。
verbosePlot:是否显示绘图的详细信息。
tmyPal:共识矩阵的可选颜色特征向量。
ml:先前的结果,如果提供则只做图形和表格。
title:输出目录的字符值。仅当 plot 不为 NULL 或 writeTable 为 TRUE 时才创建目录。此标题可以是绝对路径或相对路径。
plot:字符值。NULL – 打印到屏幕,用于位图 png 的 ‘pdf’、’png’、’pngBMP’,有助于大型数据集。
writeTable:TRUE – 将输出和日志写入 csv。
weightsItem:用于抽样项目的权重。
weightsFeature:用于抽样特征的权重。
res:consensusClusterPlus 的结果。
innerLinkage:用于子采样的分层链接方法。
finalLinkage:共识矩阵的层次链接方法。
接下来就让小师妹以GEO数据库中的GSE10245数据集为例带你使用ConsensusClusterPlus,该数据集包含了人类肺癌组织和正常肺组织的基因表达数据。
#导入数据library(GEOquery)gse <- getGEO("GSE10245")data <- exprs(gse[[1]])#数据预处理,进行数据标准化library(limma)data <- normalizeBetweenArrays(data, method="quantile")#导入ConsensusClusterPlus包,对数据进行聚类library(ConsensusClusterPlus)ccp <- ConsensusClusterPlus(data, maxK=5, reps=100, distance="pearson", clusterAlg="km")
接下来我们就可以得到8张聚类结果的图片,分别是:
四种分类的一致性矩阵和颜色的标识

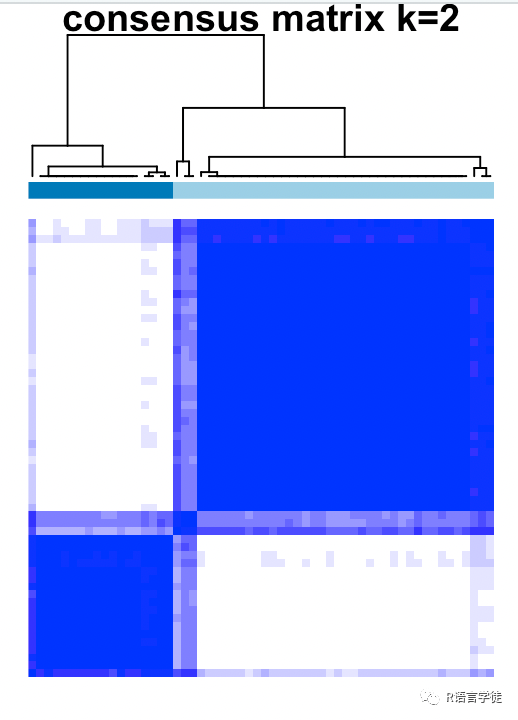


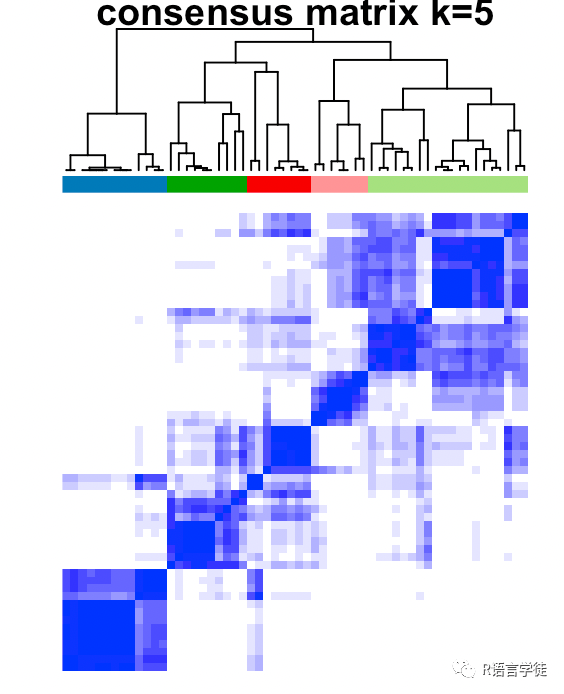
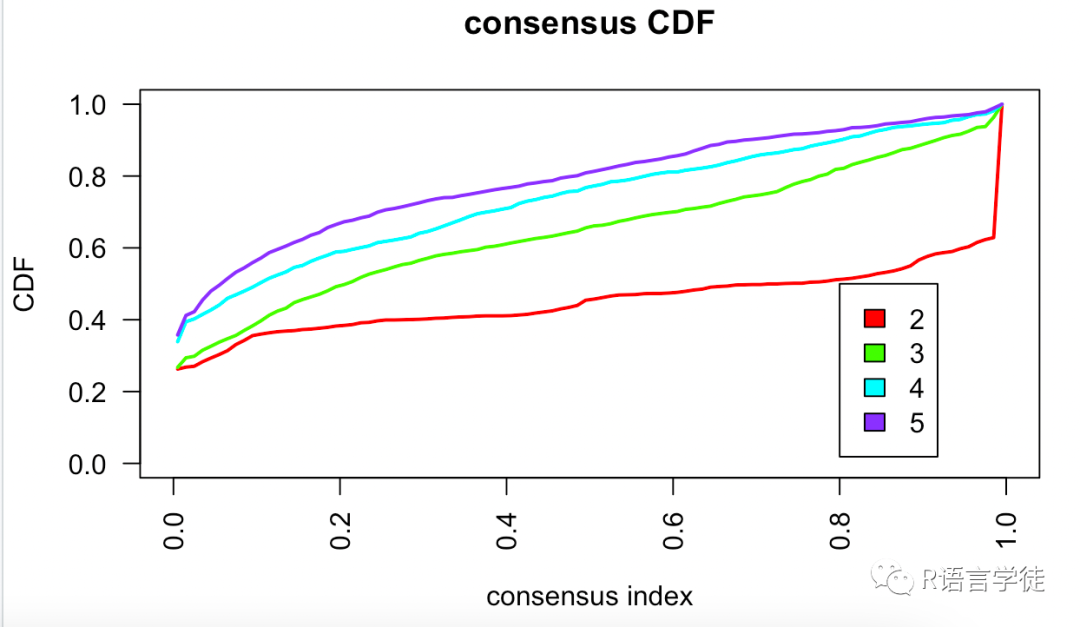
一致性累积分布函数图
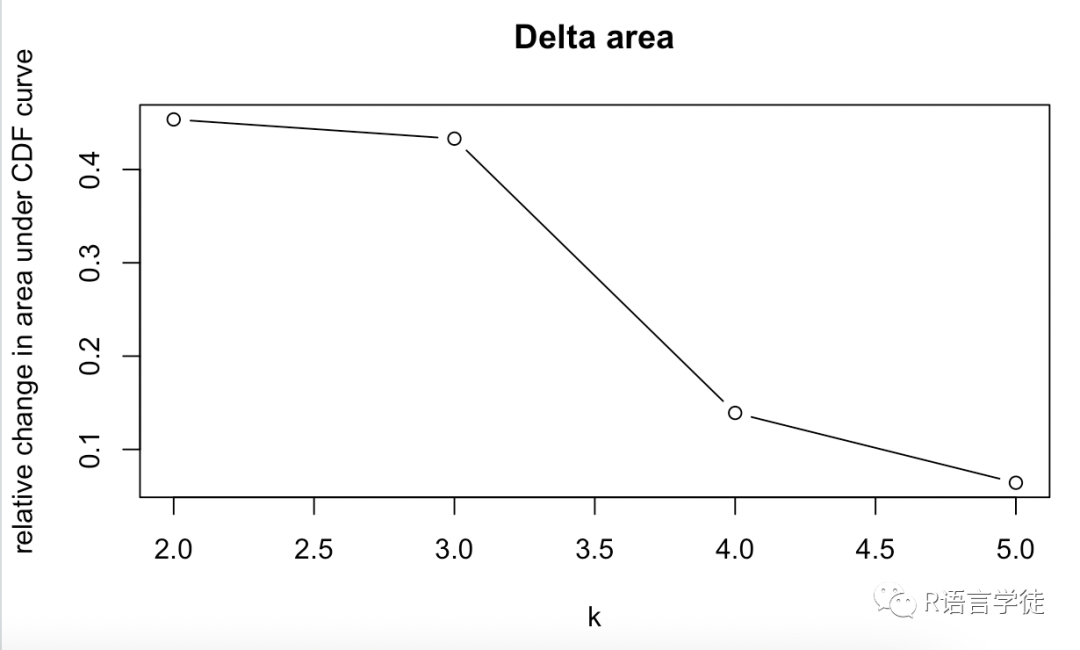
碎石图
跟踪图
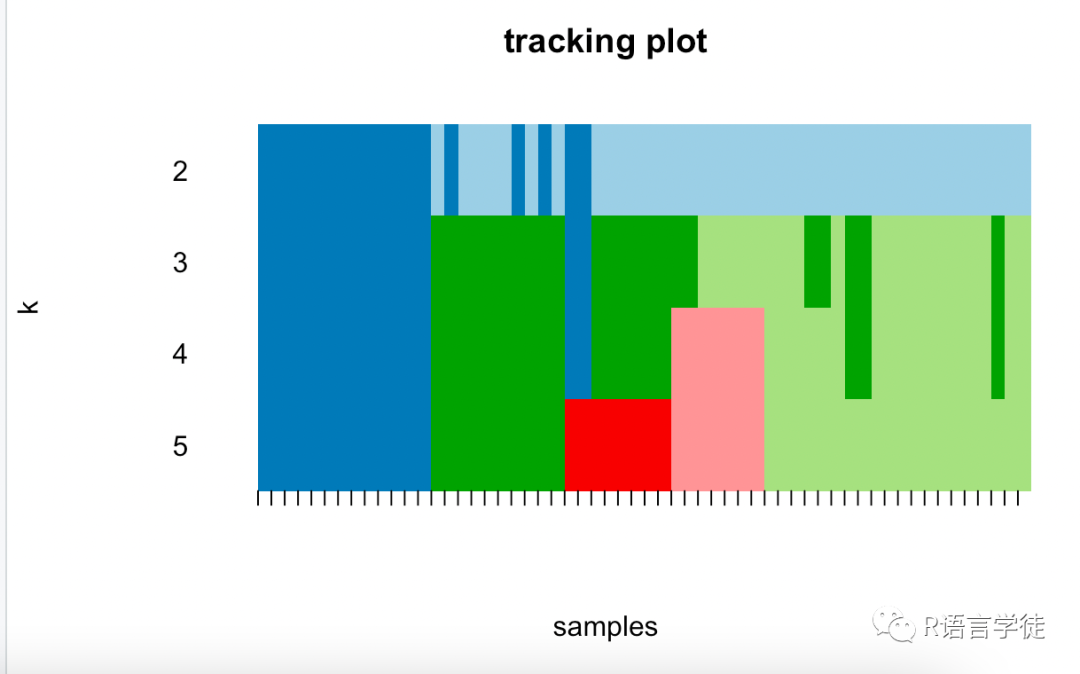
接下来使用calcICL函数计算聚类一致性和样品一致性
icl <- calcICL(ccp)
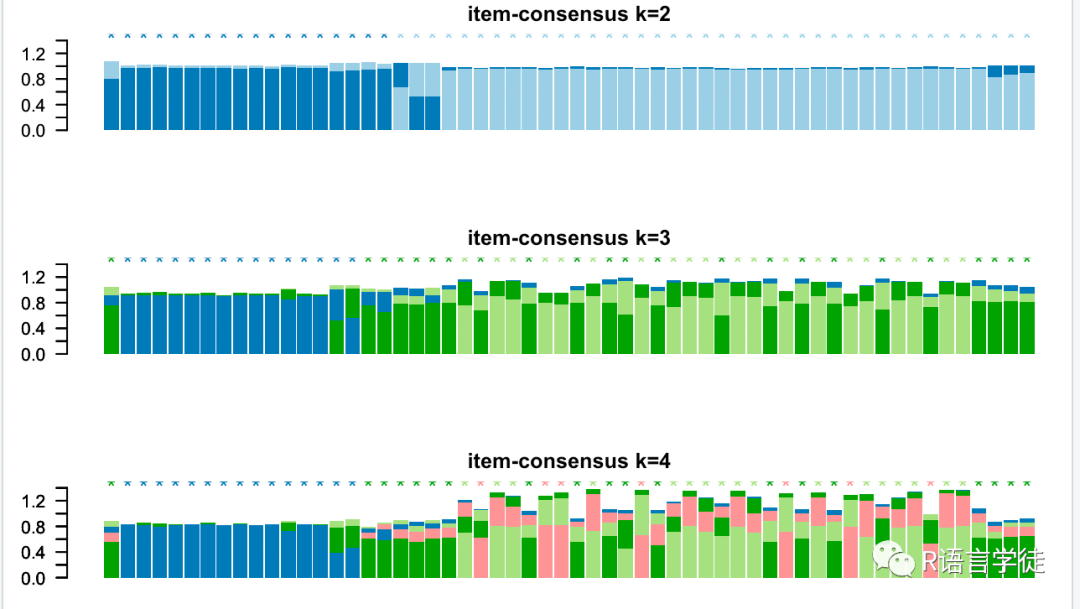
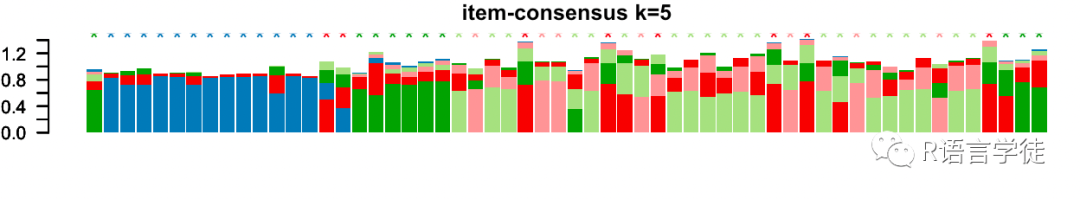
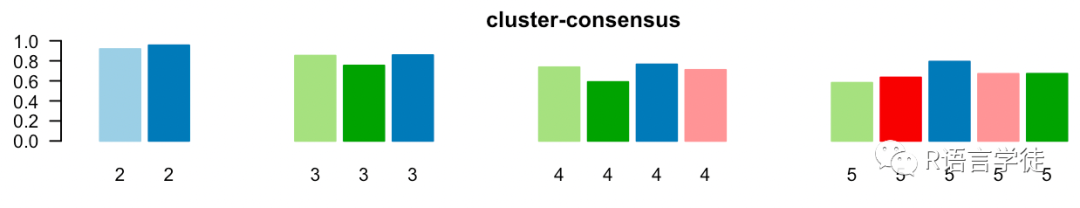
ConsensusClusterPlus短短两行代码就能带来聚类分析和14张可视化图片,是不是很神奇呢?相信也有许多小伙伴对这14张图的感到困惑,下一期小师妹将给大家细细道来每张图的意义~敬请期待~
更多实用方便的小工具在云生信平台等着你哦
http://www.biocloudservice.com/home.html
E
N
D
