一步步教你绘制GWAS分析中PCA图,曼哈顿图,QQ图
{ 点击蓝字,关注我们 }
GWAS通过分析case/control组之间的差异来寻找与疾病关联的SNP位点,然而case和control两组之间,可能本身就存在一定的差异,会影响关联分析的检测。
Population stratification,称之为群体分层,是最常见的差异来源,指的是case/control组的样本来自王不同的祖先群体,其分型结果自然是有差异的。GWAS分析的目的是寻找由于疾病导致的差异,其他的差异都属于系统误差,在进行分析时,需要进行校正。对于群体分层的校正,通常采主成分分析的方法,即PCA,主成分分析(Principal Components Analysis, PCA)是一种常用的数据降维方法,在群体遗传学中被广泛用于识别并调整样本的群体分层问题。群体分层会导致GWAS研究中的虚假关联,考虑一个case-control研究
【使用plink的得到PCA结果,一个解释多少变量,另一个是主成分分析每一个成分的数值】
#给小伙伴上代码pca_eigenvectors<-read.table('2-data-holstein_pca.eigenvec')pca_eigenval<-read.table('2-data-holstein_pca.eigenval')#calculating percentage of eigenvaluespca_eigenval$percentage<-pca_eigenval$V1/sum(pca_eigenval$V1)#plotpca_value1<-paste("PCA1(",100*round(pca_eigenval$percentage[1],3),"%)",sep = "")pca_value2<-paste("PCA2(",100*round(pca_eigenval$percentage[2],3),"%)",sep = "")# output plottiff("2-plot-PCA.tiff", width = 1000,height=1000,res = 100)par(mar=c(5,5,5,5),cex.axis=2,cex.lab=2, cex.axis=2, lwd=2 ,bty="l")plot(pca_eigenvectors[,c(3,4)],pch=16,col=rainbow(100),xlab=pca_value1,ylab=pca_value2)dev.off()
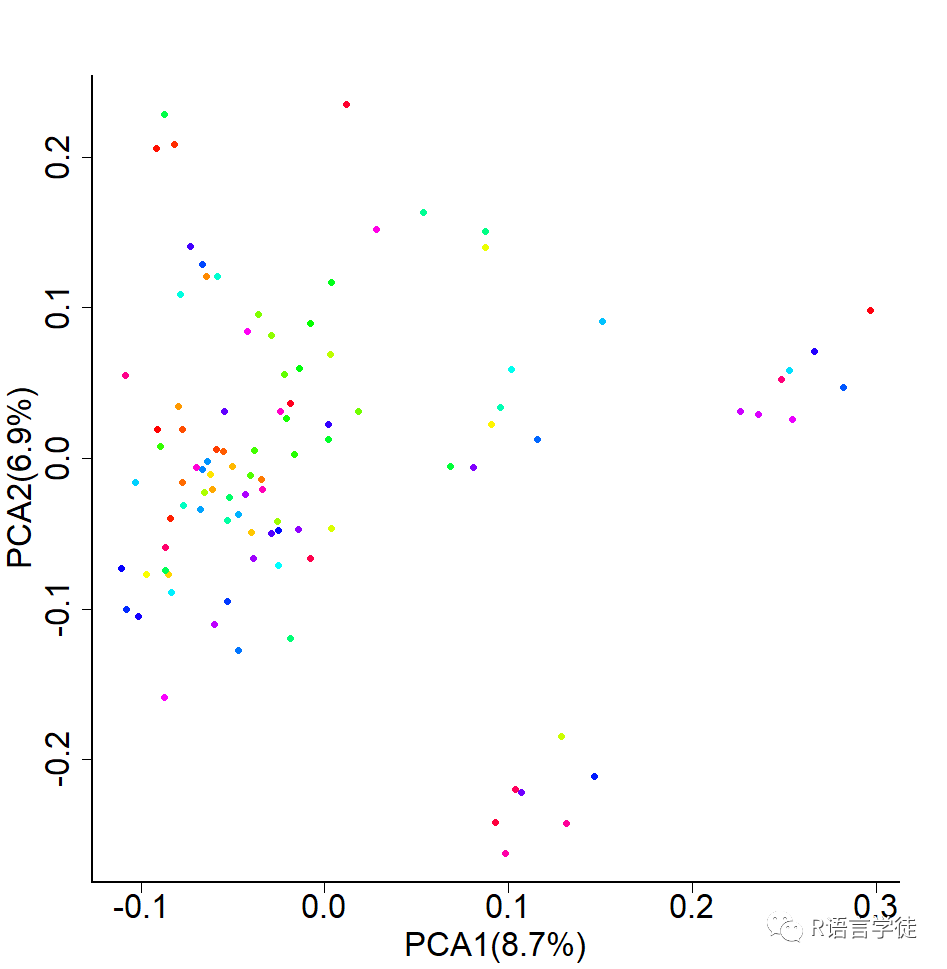
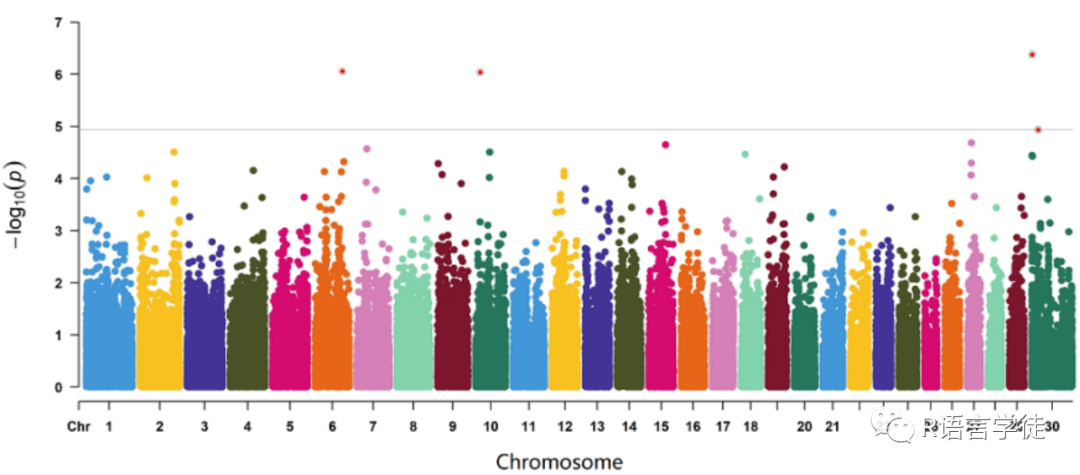
GWAS分析中,会有一个结果,每个SNP的P值,可以根据这个值,以及SNP的染色体和物理位置,进行作图。图好做,但是怎么看?怎么解读?小师妹给小伙伴解释一下。
曼哈顿图
Manhattan plot(曼哈顿图)比较简单,它是把GWAS分析之后所有SNP位点的p-value在整个基因组上从左到右依次画出来。并且,为了可以更加直观地表达结果,通常都会将p-value转换为-log10(p-value)。这样的话,基因位点-log10(p-value)在Y轴的高度就对应了与表型性状或者疾病的关联程度,关联度越强(即p-value越低)就越高。而且,一般而言,由于连锁不平衡(LD)关系的原因,那些在强关联位点周围的SNP也会跟着显示出类似的信号强度,并依次往两边递减。
本期给大家介绍R包qqman绘制曼哈顿图和QQ图
library(qqman)##输入结果文件##需要提前准备一个GWAS的结果文件,五列##第一列,snp号##第二列,snp染色体##第三列,snp位置##第四列,p值##第五列,z-得分g1=read.table("gwas_results")##读入GWAS结果文件g1=gwasResults##使用案列数据head(g1)##查看案列文件as.data.frame(table(gwasResults$CHR))##查看每条染色体分布的snp数目manhattan(gwasResults)##画出曼哈顿图##设置参数,##设置题目:main=“题目”##y周设置,ylim=c(0:10),表示y周从0到10##点的大小,cex=0.6,表示点的大小缩小60%,##x周数字,cex.axis = 0.9,x轴缩小90%##颜色,col,设置颜色##建议线,suggestiveline, genomewidelinemanhattan(gwasResults, main = "Manhattan Plot", ylim = c(0, 10), cex = 0.6,cex.axis = 0.9, col = c("blue4", "orange3"), suggestiveline = F, genomewideline = F,chrlabs = c(1:20, "P", "Q"))阈值线 abline(h=6.98,col='red') #h=-log(0.05/snp)##只花一条染色体的图manhattan(subset(gwasResults, CHR == 1))【实例操作】setwd('E:\数据\dsshh-bw-pk')g2=read.table("dsshhbw-pk.assoc.txt",header = TRUE)install.packages('qqman')library(qqman)qq1=qq(g2$p_wald,cex = 0.8)gwasResults<-cbind(g2[,c(2,1,3,12)])colnames(gwasResults) <- c("SNP","CHR","BP","P")manhattan(gwasResults, main = "Manhattan Plot", ylim = c(0, 10), cex = 0.6,cex.axis = 0.9, col = c("blue4", "orange3"), suggestiveline = F, genomewideline = F,chrlabs = c(1:27, "P", "Q"))abline(h=6.98,col='red')
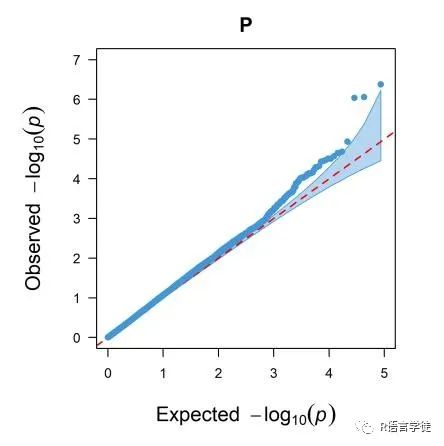
QQ图
【什么是QQ图】
QQ图,全称quantile-quantile plot,它是判断模型假阳性、假阴性的重要指标。
【为何要用QQ图来表示GWAS的结果呢】
一般,我们认为,P值达到显著性,那就说明不同的SNP分型,对表型数据是有显著性影响的。
在GWAS分析里面,QQ-plot所用的数据和上面曼哈顿图的一样,的纵轴是SNP位点的p-value值(这是实际得到的结果,observed),与曼哈顿图一样也是表示为 -log10(p-value);横轴是则是均匀分布的概率值(这是Expecte的结果),同样也是换算为-log10。
##qq图,就是看p值拟合曲线,所以提取p值qq1=qq(g1$P)#提取p值,画图head(g1)qq2=qq(g1[,4])##提取p值,画图,两个图一样
【实例操作】
setwd('F:\桌面\dsshh-bw-k')g1=read.table("dsshhbw.assoc.txt",header = TRUE)install.packages('qqman')library(qqman)qq1=qq(g1$p_wald)
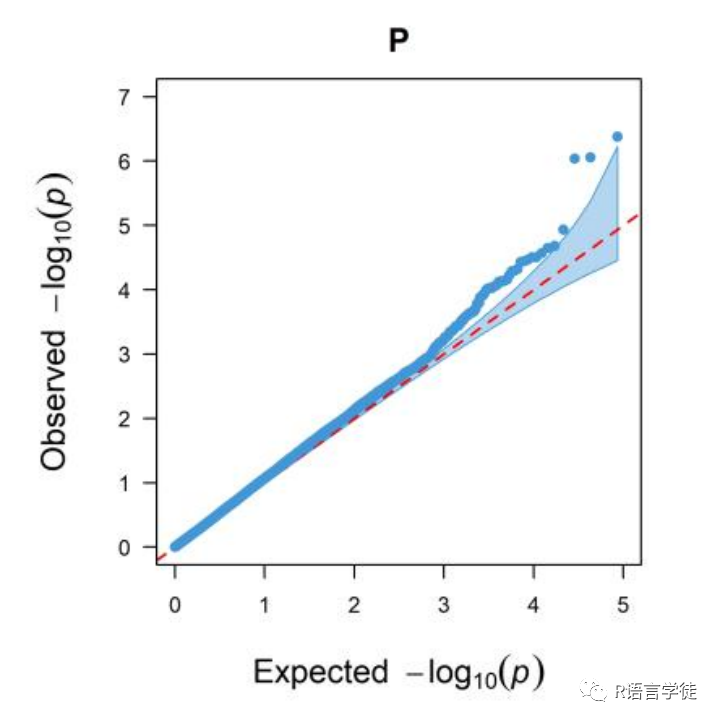
本期给大家介绍了用于展示GWAS结果曼哈顿图和QQ图的R包qqman,qqman的用法比较简单,宜上手,推荐大家使用!
如果喜欢更多的生物信息和组学文章,欢迎搜索并关注小师妹的微信公众号(R语言学徒)和云生信生物信息学平台( http://www.biocloudservice.com/home.html)
E
N
D
