保姆级教程—二代测序基因组数据的组装及基因预测

@
点击蓝字
关注我们
随着高通量测序技术的发展,二代测序如今越来越普及,测序费用也逐渐降低,因此很多研究开始使用二代测序技术来进行基因组测序,从而开展相关研究。比如在系统分类学中,由于基因组等组学数据包含更多的位点信息,基于基因组等组学数据开展的系统发育基因组学分析可以有效地消除基因水平转移和不同进化率对系统发育的影响。因此,近年来基因组数据越来越多的被用于重建系统发育树,来厘清一些存在争议的系统发育关系。但是对于一些生信小白来说,当拿到二代测序的基因组原始数据后却不知道到如何对其进行处理。今天小花就给大家带来一套从原始数据到基因预测的保姆级教程。
以下分析中所用到的软件都可以用conda安装,比如fastqc:
conda install -c bioconda fastqc
第一步:对原始数据进行质量控制
当我们拿到测序下机的原始数据后,需要对它进行一个质控,虽然测序公司一般都会对数据进行一个预处理,但是为了保证质量,我们自己还是需要进行一遍质量控制。
①首先使用fastqc查看原始数据的质量情况,结果可查看输出文件夹中的html格式文件:
fastqc -o outputdir input.fastq
-o:输出文件夹位置,需先自己在工作目录下建立一个文件夹;
input.fastq:需要分析的序列文件,可使用“*.fastq”同时分析多个原始数据。
命令示例:
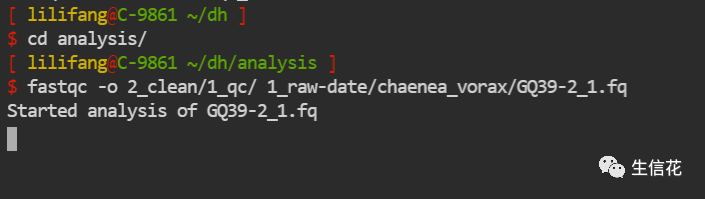
结果文件内容较多,大家可以参考:生信软件 | FastQC (测序数据质控) – 知乎 (zhihu.com)
② 经过上述步骤,我们对自己的数据质量有了一个大概了解,如果质量较好就可以直接开始组装了,要是质量不太好,我们就需要对其进行处理,以去除低质量序列。目前常用的质控软件有trimmomatic、fastp、cutadapt等,今天小花这里给大家介绍使用trimmomatic进行质控的方法。
通常来说,质量控制的标准如下:
第二步:进行组装
基因组组装可以分为从头组装(De novo assembly) 和映射比对组装(mapping assembly),。从头组装是指没有参考基因组情况下对基因组序列进行拼接;;映射比对组装就是需要把测序序列和参考基因组来比对,找到序列的对应位置再进行组装。小花这里介绍的是从头组装的方法,用到的软件为SPAdes。
SPAdes不仅支持illumina测序数据,而且还可以用于Ion Torrent测序数据,PacBio测序数据、sanger数据,Nanopore。
spades.py (-k 21,33,55,77) -1 -2 -o ./output_dir -t 16 -m 800 #双端20G以上至少需要两天时间
-k:kmer数,一次可以输入多个,用逗号分隔,kmer最大为127;如果不知道,可以不输入,软件会根据序列情况自动选择。
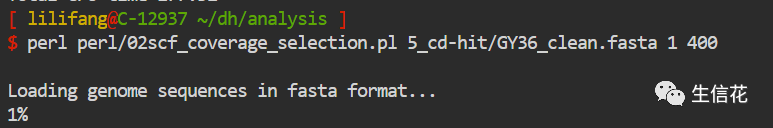
第三步:去除冗余序列
组装完后,可能有一些相似序列,这些相似序列对于后续分析会造成影响同时也会浪费一些计算资源。因此组装完后都需要进行去冗余操作。cd-hit 就是一款用于蛋白质序列或核酸序列的相似度对序列进行聚类以去除冗余序列的工具。
cd-hit-est -i scaffolds.fasta -o scaffolds_clean.fasta -c 0.98 -n 10 -r 1 -l 300 -M 40000 -T 8cd-hit:用于蛋白质序列去冗余;cd-hit-est:用于核苷酸序列去冗余-i:输入文件,组装的结果文件-o:输出文件,fasta格式,可自己命名-c:相似性,0.98表示相似性大于等于98%的为一类,并进行去冗余-n:两两序列进行序列比对时选择的 word size-M:内存大小,40000=40GB-T:使用的线程数
第四步:评估组装质量
去除冗余序列后,我们就得到了基因组序列了,我们可以用QUAST评估一下组装质量,主要是对GC含量,N50,contigs数量等进行统计。

第五步:基因预测
对于一些没有参考基因组的序列来说,要想得到蛋白质序列用于后续直系同源基因鉴定,功能注释等分析的话,就需要进行从头预测。Augustus就是一款用于从头预测的软件,该软件内部包含了一些类群的基因预测模型(可以用augustus –species=help查看),可以用来直接对自己的物种进行基因预测。
这里以拟南芥为例:
augustus --species=arabidopsis test.fa > test.gff当然对于一些比较小众的物种,可能没有被训练过的物种,要想对其进行基因预测,就需要我们先进行模型训练,再进行预测。具体步骤如下:
首先是要准备一种已知物种的核酸序列和氨基酸序列,该物种最好与要预测物种的亲缘关系要近,同属的是最好的。
1、建立连接关系
ln -s <基因.fasta> genome.faln -s <蛋白质.fasta> proteins.aa

2、运行scipio(下载地址 )制作基因结构文件
perl 2_scipio-1.4/scipio.1.4.1.pl --blat_output=prot.vs.genome.psl 1_refegene/genome.fa 1_refegene/proteins.aa > 3_traning/scipio.yaml
3、从scipio.yaml中提取GFF (General Feature Format) 基因结构文件
cat 3_training/scipio.yaml | perl 2_scipio-1.4/yaml2gff.1.4.pl > 3_training/scipio.scipiogff
perl /opt/augustus**/scripts/scipiogff2gff.pl --in=scipio.scipiogff --out=scipio.gff
4、将GFF文件转化成Genbank格式的文件
perl /opt/augustus**/scripts/gff2gbSmallDNA.pl scipio.gff genome.fa 1000 genes.raw.gb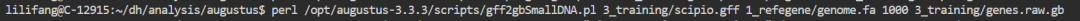
5、去除不适合trainning的基因序列
etraining --species=generic --stopCodonExcludedFromCDS=true genes.raw.gb 2> train.err
printf("hello world!");
perl /opt/augustus**/scripts/filterGenes.pl badgenes.lst genes.raw.gb > genes.gb
grep -c "LOCUS" genes.raw.gb genes.gb #(查看处理后基因序列情况)
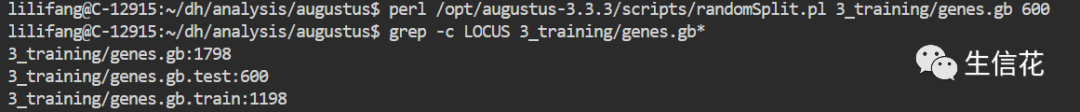
6、将用于Trainning的基因结构信息随机分为Trainning set和test set
perl /opt/augustus**/scripts/randomSplit.pl genes.gb 600
为了满足test的统计学意义,test set必须足够大,才能起到检测作用,这里取600
grep -c LOCUS genes.gb* #(查看划分后各自的基因数量)
7、在物种库添加新物种
export AUGUSTUS_CONFIG_PATH="/usr/local/augustus/config"perl /opt/augustus**/scripts/new_species.pl --species=<新物种名>

编辑bug_parameters.cfg文件,将stopCodonExcludedFromCDS 改为 true(“i”进入编辑模式,修改完成按“Esc”退出编辑模式,“:wq”+回车保存退出)

8、training
printf("hello world!");
9、测试
augustus --species=<新物种名> genes.gb.test | tee firsttest.out
grep -A 22 Evaluation firsttest.out #查看预测结果
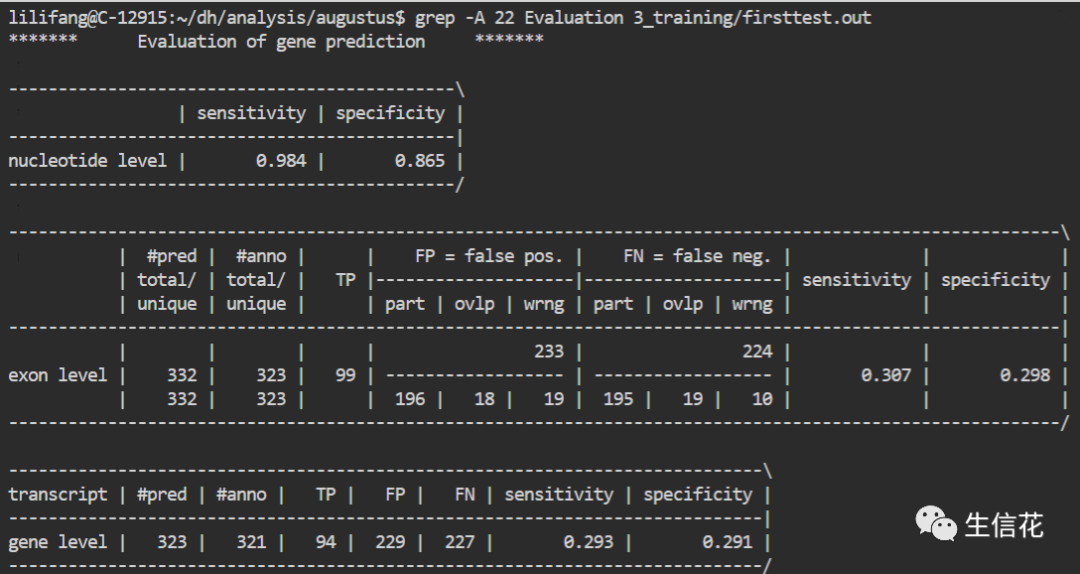
gene level的sensitivity至少大于20%,如果灵敏度不高可以用以下步骤自动调整。
10、自动优化(可选,速度慢),完成后需按7、8操作再进行一遍训练,否则无用
perl /opt/augustus-3.3.3/scripts/optimize_augustus.pl --species=litonotus_sp 3_training/genes.gb.train
11、对自己的序列进行基因预测
augustus --species=<新物种名> (input.fasta) > (output.gff)
12、提取预测结果
perl /usr/local/augustus/scripts/getAnnoFasta.pl GQ33.gff
好了,基因组从原始数据到基因预测分析的原过程到此就结束了。可以说是很详细了,不知道大家学会了吗?有什么问题都可以和小花来讨论哦!我们下期再见!
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
提醒:以上分析中有一些软件比较耗时,且对电脑内存等性能要求比较高,推荐大家用服务器。如果大家想用服务器可以联系微信:18502195490(快来联系我们使用吧!)


(点击阅读原文跳转)
![]() 点一下阅读原文了解更多资讯
点一下阅读原文了解更多资讯
