大海哥带你绘制最简单的基因表达的泛癌分析
点击蓝字 关注我们
泛癌分析是对多种癌症类型进行整体分析和比较的研究方法。综合考虑多种癌症类型的共性和差异,揭示出癌症发展的一般性规律和生物学机制。发现潜在靶。
对多个癌症类型的基因表达、突变、融合等信息进行综合分析,可以发现共同的异常信号和靶标,这些靶标可能在多种癌症类型中起到重要作用。这有助于发现潜在的治疗靶点和预后标志物,提供更广泛的治疗选择和个体化治疗策略。
识别癌症驱动基因:泛癌分析可以帮助识别在多种癌症类型中高度共有的癌症驱动基因。这些基因对于癌症发展和进展起到关键作用,因此成为重要的治疗目标和研究对象。通过对这些基因的深入研究,可以更好地理解癌症的发生机制,并为癌症治疗提供新的靶向治疗策略。
箱线图(Boxplot)是一种常用的统计图形,用于显示数据集的分布特征和离群值情况。它通过五个统计量(最小值、下四分位数、中位数、上四分位数、最大值)来描述数据的位置和离散程度。
下面来看代码,这是示例数据。
#install.packages("ggpubr")library(ggpubr) #引用包library(ggpubr)加载ggpubr包,该包提供了用于绘制箱型图和统计分析的函数。inputFile="geneExp.txt" #输入文件名称setwd("C:\biowolf\geneCeRNA\08.panDiff") #使用setwd()函数设置工作目录,即数据文件所在的目录
#读取输入文件data=read.table(inputFile, header=T, sep="t", check.names=F)
#读取基因表达数据文件,并根据癌症类型进行分组,然后绘制箱型图来展示不同癌症类型中基因表达的差异。gene=colnames(data)[2]colnames(data)[2]="expression"##gene变量存储基因名,colnames(data)[2]将第二列的列名(基因表达值)更改为"expression"。统计每个肿瘤正常样品数目:从数据框中选择正常样品,并使用table()函数统计每个肿瘤类型的样品数目。NormalNum保存样品数目大于等于5的肿瘤类型。NormalCancer保存样品数目大于等于5的肿瘤类型的名称。数据筛选:使用which()函数和%in%操作符,从数据框中选择属于NormalCancer中肿瘤类型的数据。#统计每个肿瘤正常样品数目,挑选正常样品数目>=5的肿瘤Normal=data[data[,"Type"]=="Normal",]NormalNum=table(Normal[,"CancerType"])NormalNum=NormalNum[NormalNum>=5]NormalCacner=names(NormalNum)data=data[which(data[,"CancerType"] %in% NormalCacner),]#绘制箱型图p=ggboxplot(data, x="CancerType", y="expression", color="Type",xlab="",ylab=paste0(gene," expression"),palette = c("blue","red") )p=p+rotate_x_text(60)#绘制箱型图:使用ggboxplot()函数绘制箱型图。x="CancerType"指定x轴为癌症类型,y="expression"指定y轴为基因表达值。color="Type"指定根据样品类型(正常/肿瘤)进行颜色区分。xlab=""将x轴标签设置为空,ylab=paste0(gene," expression")设置y轴标签为基因名 + "expression"。palette = c("blue","red")设置颜色方案,蓝色表示正常样品,红色表示肿瘤样品。rotate_x_text(60)将x轴标签旋转60度,以避免标签重叠。#输出图形pdf(file="boxplot.pdf", width=7.5, height=5)p+stat_compare_means(aes(group=Type),method="wilcox.test",#统计分析方法(wilcox.test)来比较不同类型之间的差异symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", " ")),#图中标注显著性水平label = "p.signif")dev.off()###设置输出图形的大小。stat_compare_means()函数于在箱型图上进行统计分析,标注不同癌症类型之间的显著性差异。aes(group=Type)指定进行比较的分组变量为样品类型(正常/肿瘤)。method="wilcox.test"使用Wilcoxon秩和检验进行比较。symnum.args参数定义了显著性水平的符号表示。label="p.signif"标注显著性水平。dev.off()关闭pdf输出。
来给大家展示展示结果图吧,使用firefox命令。
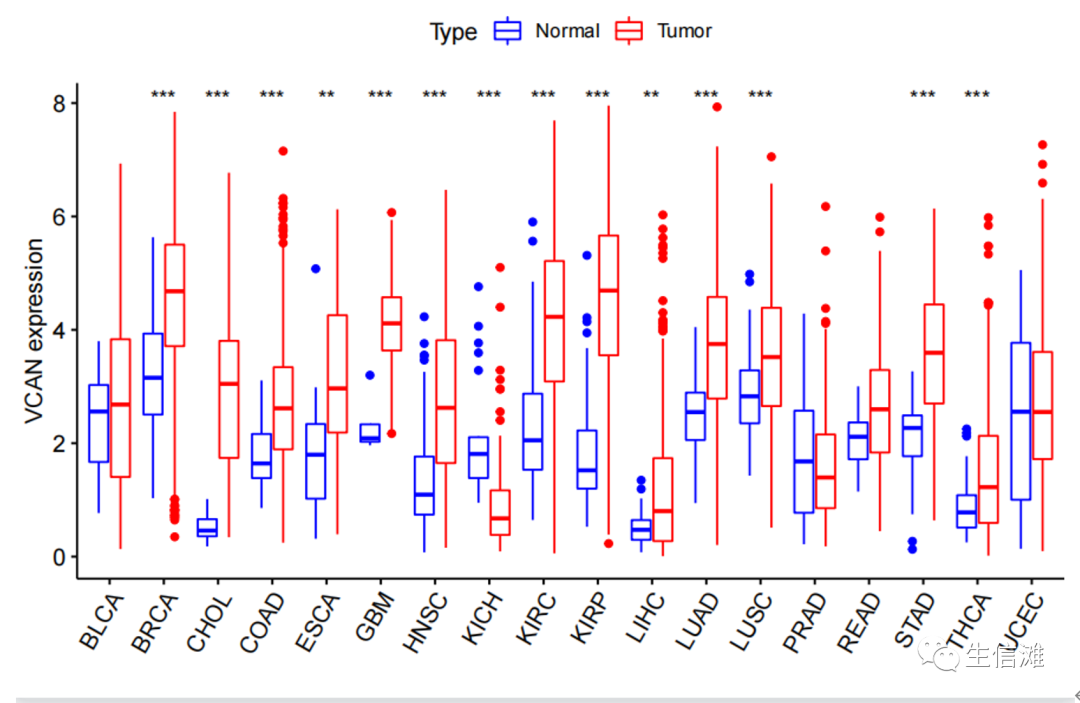
根据基因表达数据绘制箱型图,展示不同癌症类型中基因表达的差异,并通过统计分析方法比较不同癌症类型之间的显著性差异。这可以帮助了解不同癌症类型中基因表达的变化,并发现可能与癌症发展相关的基因。
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:
http://www.biocloudservice.com/home.html


点击“阅读原文”进入网址