保姆级带你代码解决单基因泛癌富集分析
点击蓝字,关注我们
我们对给定的肿瘤数据进行基因集富集分析,以寻找与指定基因(例如BRCA1)相关的生物学功能和通路。这对于深入理解基因在不同肿瘤类型中的功能以及与肿瘤发生和发展相关的生物学过程非常重要。基因集富集分析是一种常用的生物信息学方法,它可以通过比较基因集内的基因在样本中的表达模式,找出与该基因集相关联的生物学过程、通路或功能。这种分析为研究人员提供了在高通量数据中寻找生物学意义的有力工具,有助于识别潜在的靶点、生物标志物以及开展更深入的机制研究。
代码中对选定的基因brca1表达量进行富集分析。下面是下载的GO和KEGGgmt文件,方便后续使用R包”org.Hs.eg.db”、”DOSE”、”clusterProfiler”和”enrichplot”分析。


#install.packages("colorspace")#install.packages("stringi")#install.packages("ggplot2")安装三个R包:"colorspace"、"stringi"和"ggplot2"。这些包可能是后续代码运行所需的依赖包。#if (!requireNamespace("BiocManager", quietly = TRUE))# install.packages("BiocManager")#BiocManager::install("limma")#BiocManager::install("org.Hs.eg.db")#BiocManager::install("DOSE")#BiocManager::install("clusterProfiler")#BiocManager::install("enrichplot")生物信息学和富集分析相关的R包,这些包包括:"BiocManager"、"limma"、"org.Hs.eg.db"、"DOSE"、"clusterProfiler"和"enrichplot"。这些包提供了进行基因集富集分析所需的函数和数据库。library(limma)library(org.Hs.eg.db)library(clusterProfiler)library(enrichplot)"library"函数加载了之前安装的四个包:"limma"、"org.Hs.eg.db"、"clusterProfiler"和"enrichplot",使得这些包的函数可以在后续代码中使用。定义了变量"gene",表示感兴趣的基因名称,以及变量"gmtFile",表示基因集文件的名称。gene="BRCA1" #基因名称gmtFile="c5.all.v7.1.symbols.gmt" #基因集文件#"read.gmt"函数从指定的基因集文件"gmtFile"中读取基因集信息,将基因集存储在变量"gmt"中。gmt=read.gmt(gmtFile)#获取目录下的所有肿瘤数据文件files=dir()files=grep("^symbol.",files,value=T)获取当前工作目录下所有以"symbol."开头的文件名,将这些文件名存储在变量"files"中。这里的文件名可能是肿瘤的数据文件,以"symbol."开头可能是因为这些文件中的基因表达数据使用基因符号(symbol)作为列名。for(i in files){#读取当前肿瘤数据文件,将数据存储在变量"rt"中。提取肿瘤类型名称,用于后续输出文件的命名。rt=read.table(i,sep="t",header=T,check.names=F)CancerType=gsub("^symbol\.|\.txt$","",i)#对基因表达数据进行预处理:如果一个基因占了多行,取其均值作为该基因的表达值。rt=as.matrix(rt)rownames(rt)=rt[,1]exp=rt[,2:ncol(rt)]dimnames=list(rownames(exp),colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)data=avereps(data)#删除正常样品:根据样本的命名规则(其中"-"分隔),#筛选出正常样品(命名中不包含"2"),并将这些样品从数据中删除。因为这里的富集分析主要关注癌症样本。group=sapply(strsplit(colnames(data),"\-"),"[",4)group=sapply(strsplit(group,""),"[",1)group=gsub("2","1",group)data=data[,group==0]#按目标基因将样品分成高低表达两组,根据指定的基因(gene)将样本分成高表达组和低表达组,#基于基因的表达量的中位数来分组。dataL=data[,(data[gene,]<=median(data[gene,]))]dataH=data[,(data[gene,]>median(data[gene,]))]meanL=rowMeans(dataL)meanH=rowMeans(dataH)meanL[meanL<0.00001]=0.00001meanH[meanH<0.00001]=0.00001logFC=log2(meanH/meanL)logFC=sort(logFC,decreasing=T)#计算基因的logFC(log2 fold change)值,#用来衡量在高表达组与低表达组之间基因表达的差异#富集分析GSEA(Gene Set Enrichment Analysis)来进行基因集富集分析。#我们通过调用GSEA函数GSEA(logFC, TERM2GENE=gmt, nPerm=100, pvalueCutoff=1)来执行富集分析,其中logFC是基因的logFC值,#TERM2GENE是基因集数据库,nPerm是随机重排的次数,pvalueCutoff是设定的p值阈值。kk=GSEA(logFC,TERM2GENE=gmt, nPerm=100,pvalueCutoff = 1)kkTab=as.data.frame(kk)kkTab=kkTab[kkTab$pvalue<0.05,]write.table(kkTab,file=paste0("Term.",CancerType,".txt"),sep="t",quote=F,row.names = F)#富集分析结果保存为一个数据框kkTab,并将p值小于0.05的富集结果筛选出来。#富集分析结果写入名为"Term..txt"的文本文件,其中CancerType是肿瘤类型。#输出富集的图形#有至少5个富集项,则输出前5个富集图形,每个图形保存为一个PDF文件,文件名为"Term..pdf",#其中CancerType是肿瘤类型。termNum=5if(nrow(kkTab)>=termNum){gseaplot=gseaplot2(kk, row.names(kkTab)[1:termNum],base_size =8)pdf(file=paste0("Term.",CancerType,".pdf"),width=8,height=6)print(gseaplot)dev.off()}}


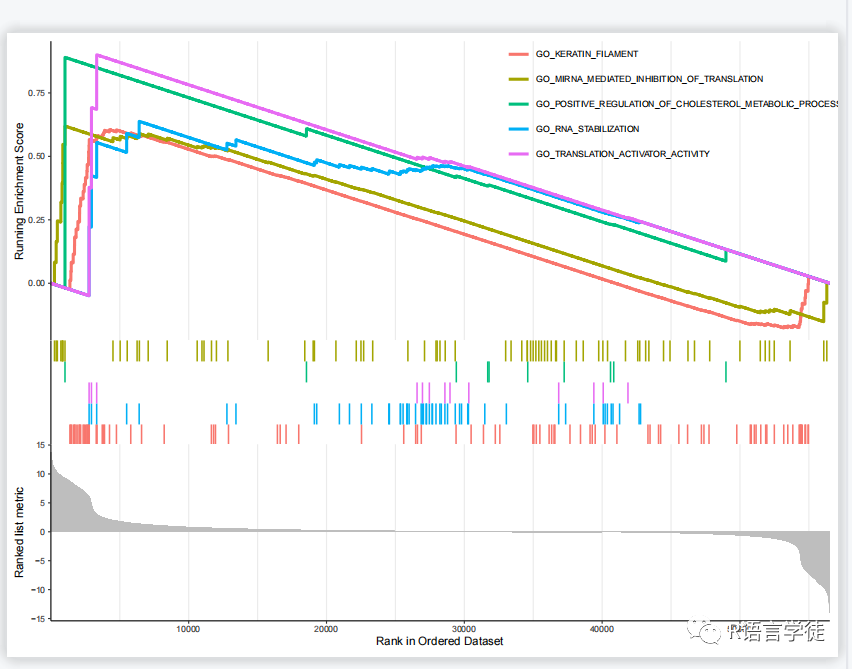
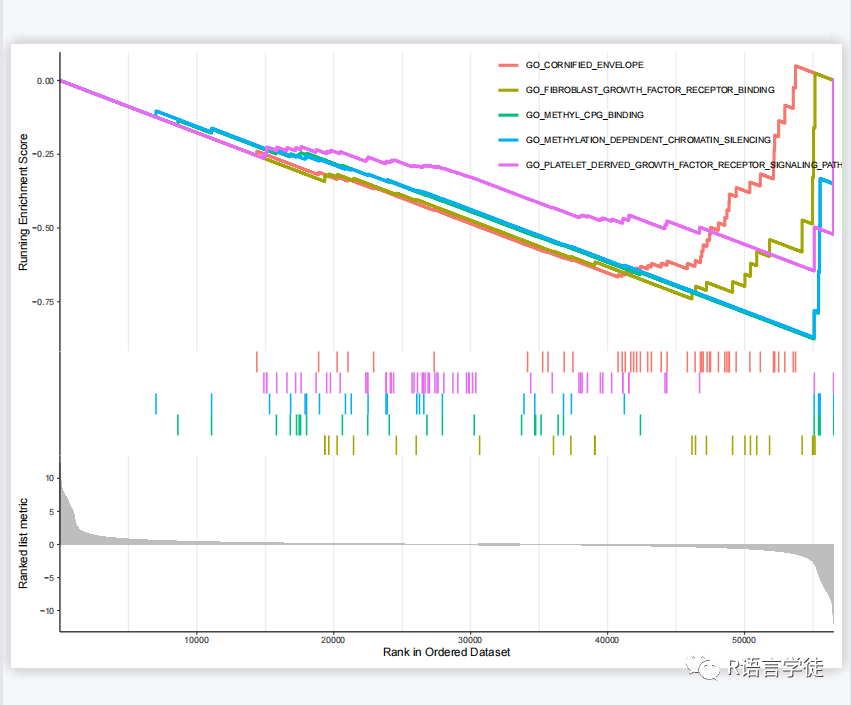
以上就是对给定的肿瘤数据,以及指定的基因集数据库(gmt文件),进行了基因集富集分析,通过输出富集分析结果的文本文件和富集图形,帮助研究人员了解在不同肿瘤类型中与指定基因相关的生物学功能和通路。
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:
http://www.biocloudservice.com/home.html。
云生信平台链接:
http://www.biocloudservice.com/home.html。
1
END
1

