拟时序分析不会做?R包Monocle3来解决
点击蓝字
关注小图
今天小图来为大家介绍细胞拟时序分析以及如何使用monocle3做细胞拟时序分析。
那么什么是细胞拟时序分析呢?
在生命体发育过程中,细胞会在特定条件下进行分化,从一个状态过渡到另一个状态,表达完全不同的基因集,有的基因被沉默,而有的基因被激活。这些状态通常很难表征,因为想要纯化中间状态的细胞很困难或不可能。但是通过单细胞测序技术捕获一群细胞的转录快照,可以使用轨迹推断(trajectory inference)的方法,对测序的细胞根据表达模式的相似性进行排序。由此一来就可以模拟细胞的动态变化。
为什么叫做伪时序分析呢?
如果想要得到细胞分化轨迹,分化的时序分析,理论上的做法是设置多个时间点,每个时间点取一次样,进行测序以及后续的分析。但是这种方法很难做,而且费时费力费钱。通过单细胞测序可以从一群细胞中得到转录瞬时快照,可以从中推断出细胞的分化轨迹。这种方法因为只取了一个时间点,所以叫做伪时序分析。

接下来小图带大家用monocle3来做伪时序分析。
一、安装monocle3
首先安装Bioconductor, 打开Rstudio并且运行
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install(version = "3.14")
接着安装一些Bioconductor不会自动安装的依赖
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats','limma', 'lme4', 'S4Vectors', 'SingleCellExperiment','SummarizedExperiment', 'batchelor', 'HDF5Array','terra', 'ggrastr'))
然后安装monocle3
install.packages("devtools")devtools::install_github('cole-trapnell-lab/monocle3')
通常这会需要一定的时间。
加载monocle3
library(monocle3)二、数据加载与预处理
小图这里使用一个秀丽隐杆线虫数据集,这一数据集来自Packer&Zhu等人。他们的研究包括对整个胚胎发育过程的时间序列分析。我们将使用数据的一小部分,其中包括大多数神经元细胞。
#加载数据expression_matrix <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_expression.rds"))cell_metadata <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_colData.rds"))gene_annotation <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_rowData.rds"))cds <- new_cell_data_set(expression_matrix,cell_metadata = cell_metadata,gene_metadata = gene_annotation)#数据预处理#数据预处理cds <- preprocess_cds(cds, num_dim = 50)cds <- align_cds(cds, alignment_group = "batch", residual_model_formula_str = "~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading")
使用alignment_group对其不同批次细胞。bg.300等每一列对应与细胞可能被污染的信号,将这些项通过residual_model_formula_str传递给align_cds,在后续的处理中减去这些背景信号。
三、降低维度并可视化结果
接下来可以对数据进行降维,monocle3建议使用默认的UMAP算法降维。
cds <- reduce_dimension(cds)plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = "cell.type")

Monocle重建了带有许多分支的轨迹
使用plot_cells()来可视化单个基因如何沿轨迹变化。让我们来看看一些在纤毛神经元中具有有趣表达模式的基因:
ciliated_genes <- c("che-1","hlh-17","nhr-6","dmd-6","ceh-36","ham-1")plot_cells(cds,genes=ciliated_genes,label_cell_groups=FALSE,show_trajectory_graph=FALSE)
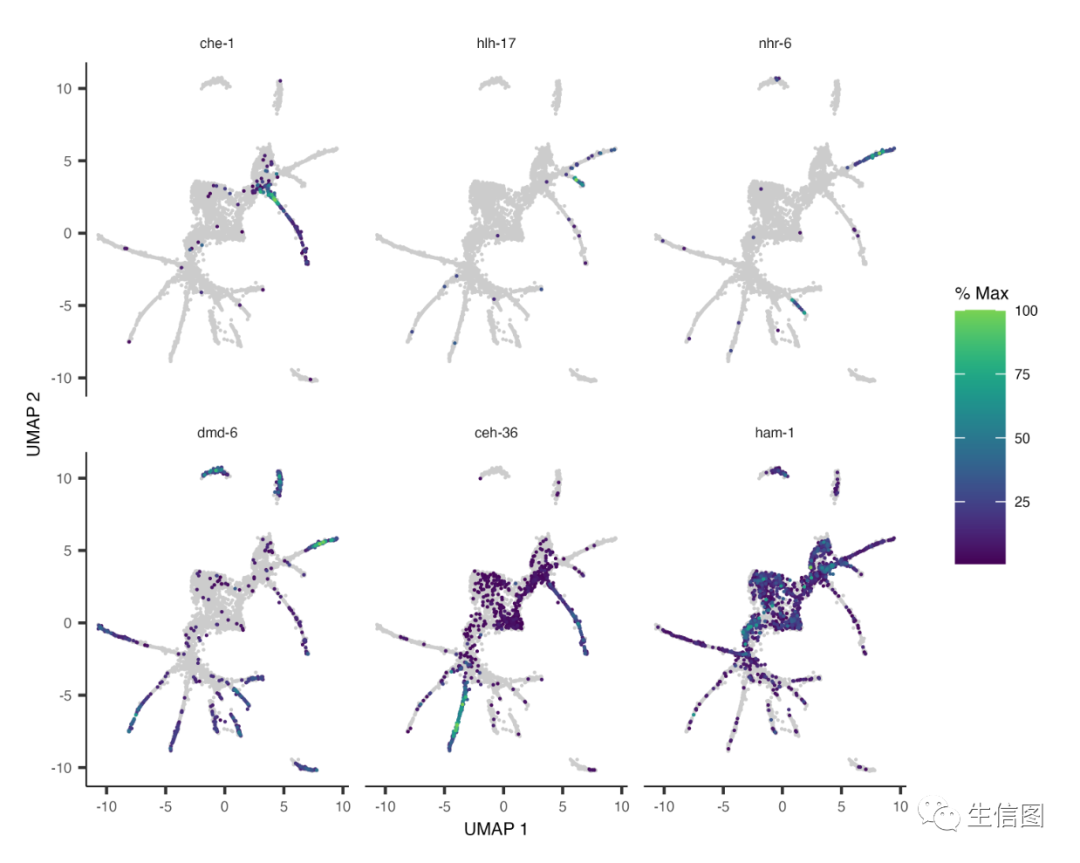
6个基因的轨迹变化
四、聚类细胞
虽然细胞可以从一种状态持续过渡到下一种状态,它们之间没有离散的边界,但是Monocle并不认为数据集中的所有细胞都来自一个共同的转录“祖先”。Monocle能够通过其聚类程序了解细胞何时应该放置在相同的轨迹中,而不是放置在不同的轨迹中。
cds <- cluster_cells(cds)plot_cells(cds, color_cells_by = "partition")

将细胞分为了四类
接下来,我们将使用最关键的Learn_graph()函数在每个分区中拟合一个主要图:
cds <- learn_graph(cds)plot_cells(cds,color_cells_by = "cell.type",label_groups_by_cluster=FALSE,label_leaves=FALSE,label_branch_points=FALSE)
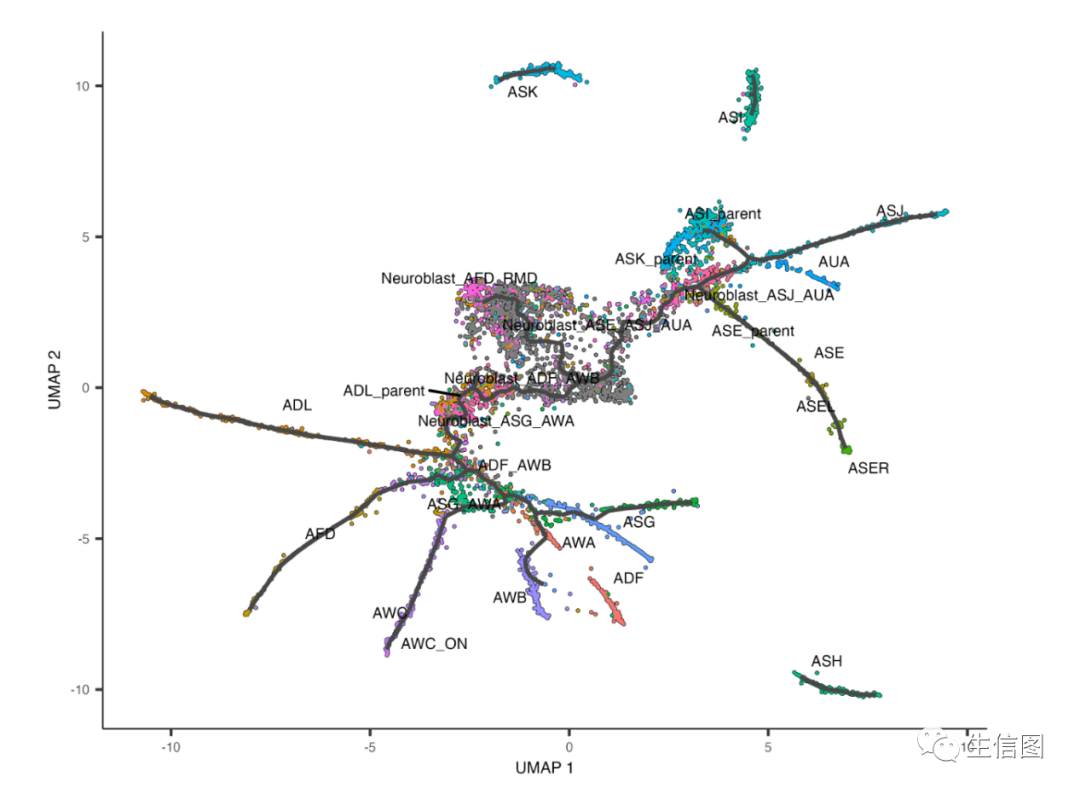
该图将用于许多下游步骤,如分支分析和微分表达式
五、细胞伪时序分析
为了将细胞排列整齐,我们需要告诉Monocle生物过程的“开始”在哪里。我们通过选择图中标记为轨迹“根”的区域来做到这一点。在时间序列实验中,这通常可以通过在UMAP空间中找到由早期时间点的细胞占据的点来实现:
plot_cells(cds,color_cells_by = "embryo.time.bin",label_cell_groups=FALSE,label_leaves=TRUE,label_branch_points=TRUE,graph_label_size=1.5)
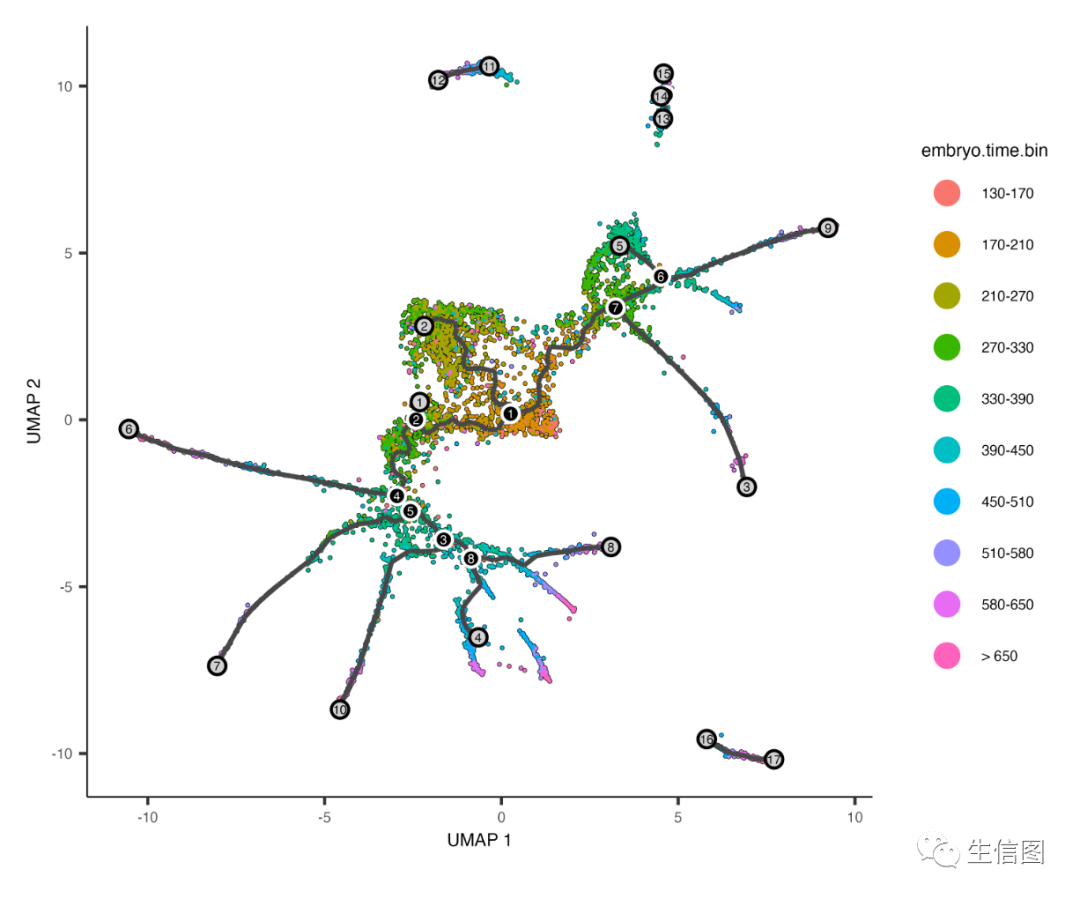
黑线显示了图的结构,带数字的圆圈表示图形中的特殊点。每片叶子,用浅灰色圆圈表示,对应于轨迹的不同结果(即细胞命运)。黑色圆圈表示分支节点,其中细胞可以传播到几个结果之一。
1
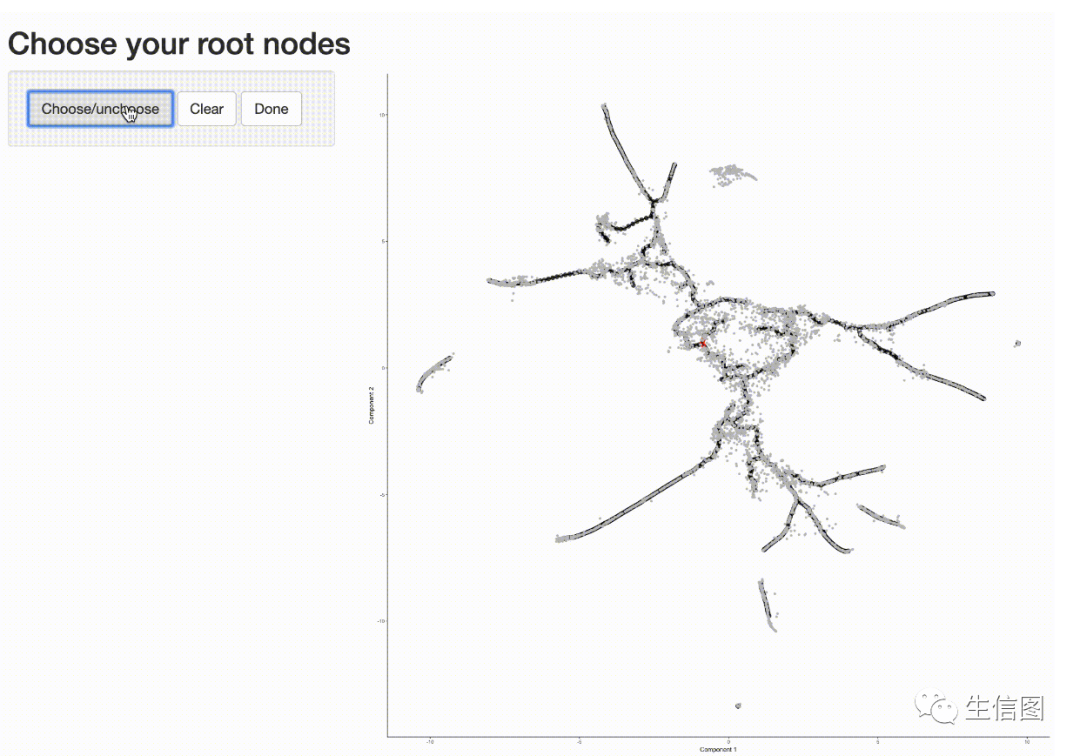
手动选取根细胞
cds <- order_cells(cds)现在我们已经知道了早期细胞的下落,我们可以调用order_cells(),它将计算每个细胞在伪时间内的下落。为了这样做,order_cells()需要您指定轨迹图的根节点。如果您不提供它们作为参数,它将启动用于选择一个或多个根节点的图形用户界面。
绘制细胞并通过假时间对其着色显示了它们是如何排列的:
plot_cells(cds,color_cells_by = "pseudotime",label_cell_groups=FALSE,label_leaves=FALSE,label_branch_points=FALSE,graph_label_size=1.5)
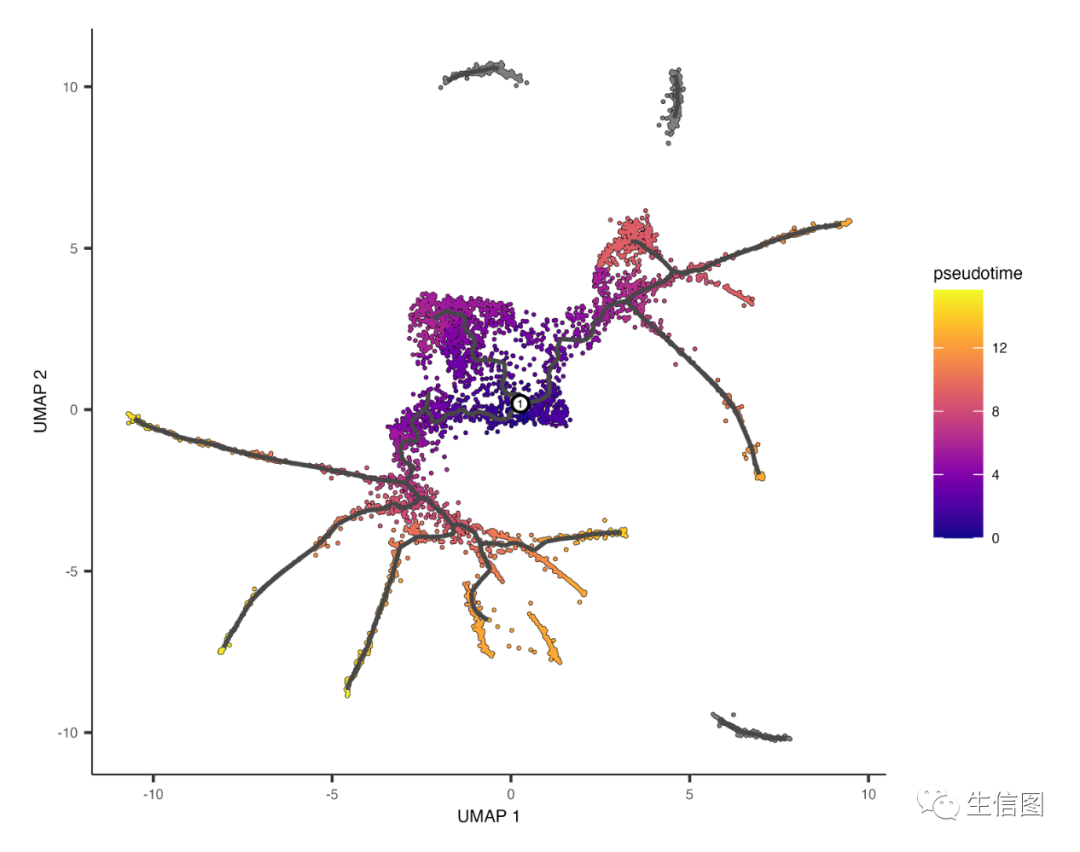
有些单元格是灰色的。这意味着它们有无限的伪时间,因为它们无法从根节点访问。
2
通过代码拾取根节点
通常需要以编程方式指定轨迹的根,而不是手动拾取。下面的函数首先根据最接近的轨迹图节点对单元格进行分组。然后,它计算每个节点的单元格中来自最早时间点的部分。然后,它选择早期细胞占用最多的节点,并将其作为根返回。
get_earliest_principal_node <- function(cds, time_bin="130-170"){cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)closest_vertex <-cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertexclosest_vertex <- as.matrix(closest_vertex[colnames(cds), ])root_pr_nodes <-igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,]))))]root_pr_nodes}
通过root_pr_node参数将程序选择的根节点传递给order_cells()会产生:
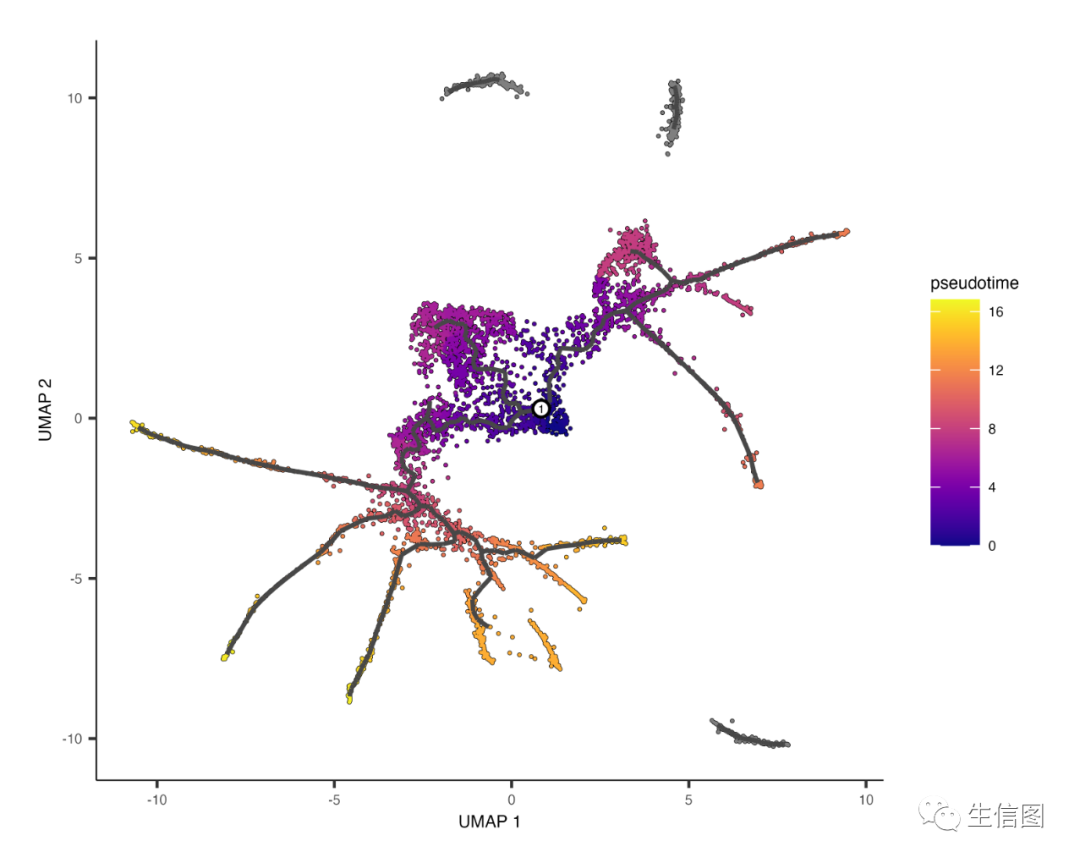
3
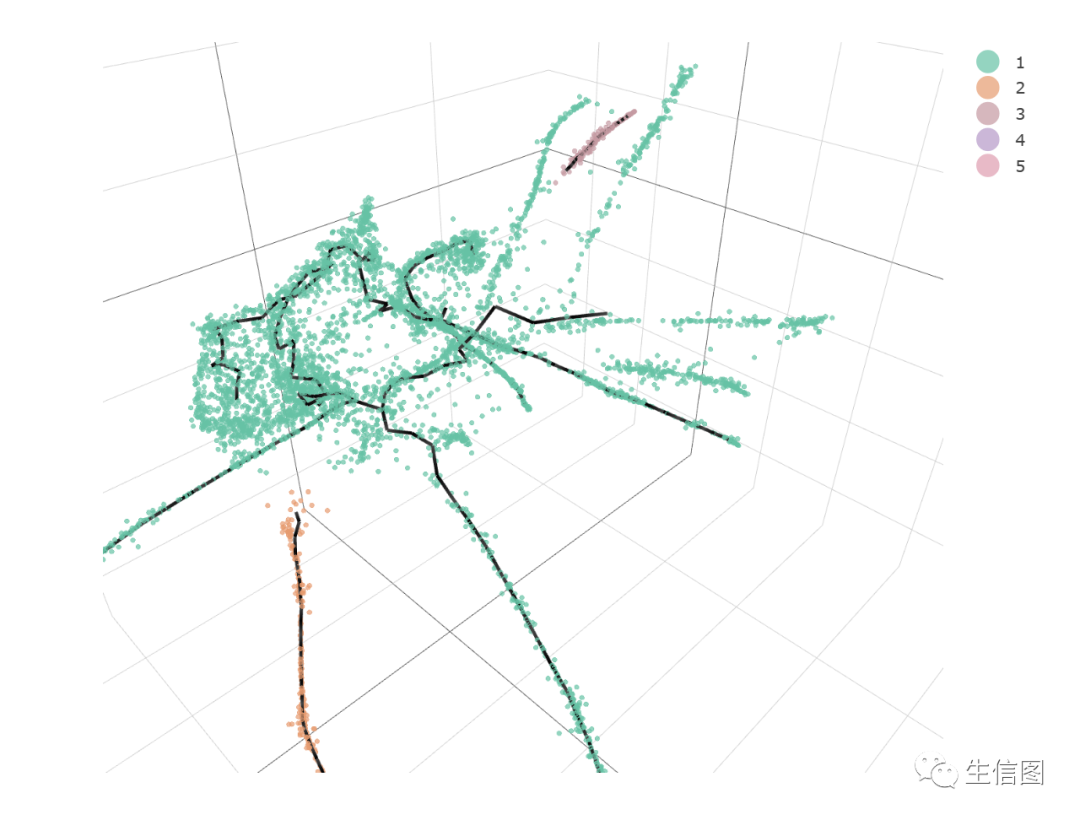
生成3D图
我们还可以生成3D的细胞分化轨迹图。
cds_3d <- reduce_dimension(cds, max_components = 3)cds_3d <- cluster_cells(cds_3d)cds_3d <- learn_graph(cds_3d)cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by="partition")
小图今天的讲解就到这里了~我们下期见。
本公司开发的云平台生物信息分析工具,零代码完成分析,同时也有许多生物处理的好用工具哦~云平台网址:http://www.biocloudservice.com/home.html
欢迎使用:云生信平台 ( http://www.biocloudservice.com/home.html)

|
往期推荐 |
|
|
|
|
|
|
👇点击阅读原文进入网址