一个宝藏工具,bwa带你玩转序列比对!

hello,你知道有一个软件包,可以能够将差异度较小的序列比对到一个较大的参考基因组上嘛?没错,它就是我们今天要学习的主角–bwa,今天小果手把手教你如何下载bwa以及如何使用它进行序列比对,感兴趣的话就和小果一起看下去吧!
公众号回复“111”领取文献,文献编号:231106
什么是bwa
BWA(Burrows-Wheeler Aligner)是一款广泛使用的生物信息学工具,用于将DNA测序数据与参考基因组进行比对。它采用了Burrows-Wheeler变换和后缀数组索引等高效算法,能够快速且准确地比对测序数据。
BWA主要包含以下三个子程序:
1.bwa index:用于构建参考基因组的索引。在比对之前,必须先对参考基因组进行索引构建。索引可以大幅提高比对的速度和效果。
2.bwa mem:是最常用的子程序,用于将测序数据与参考基因组进行比对。它适用于短读长和长读长的测序数据,能够高效地处理插入、删除和错配等多种比对情况,输出比对结果以供进一步分析。
3.bwa samse/sampe:用于将比对结果转换为SAM(Sequence Alignment/Map)格式。SAM格式是一种常见的存储比对结果的标准格式,包含了序列比对的详细信息,如比对位置、质量分数等。
BWA具有许多优势,包括速度快、准确性高、适用于不同的测序数据类型等。它被广泛应用于基因组比对、变异检测、转录组分析等生物信息学研究领域。
bwa序列比对流程
数据准备
对于今天的学习,我们需要准备同一个物种的参考基因组和待测序列数据,在这里小果以酵母的参考基因组和待测序列为例向大家演示如何使用bwa:
1.S288C_reference_sequence_R64-3-1_20210421.fna ---酵母数据参考基因组2.new4257-contigs.fa ---酵母数据待测短序列
我们一起来看一下这两个数据的文件格式:
S288C_reference_sequence_R64-3-1_20210421.fna 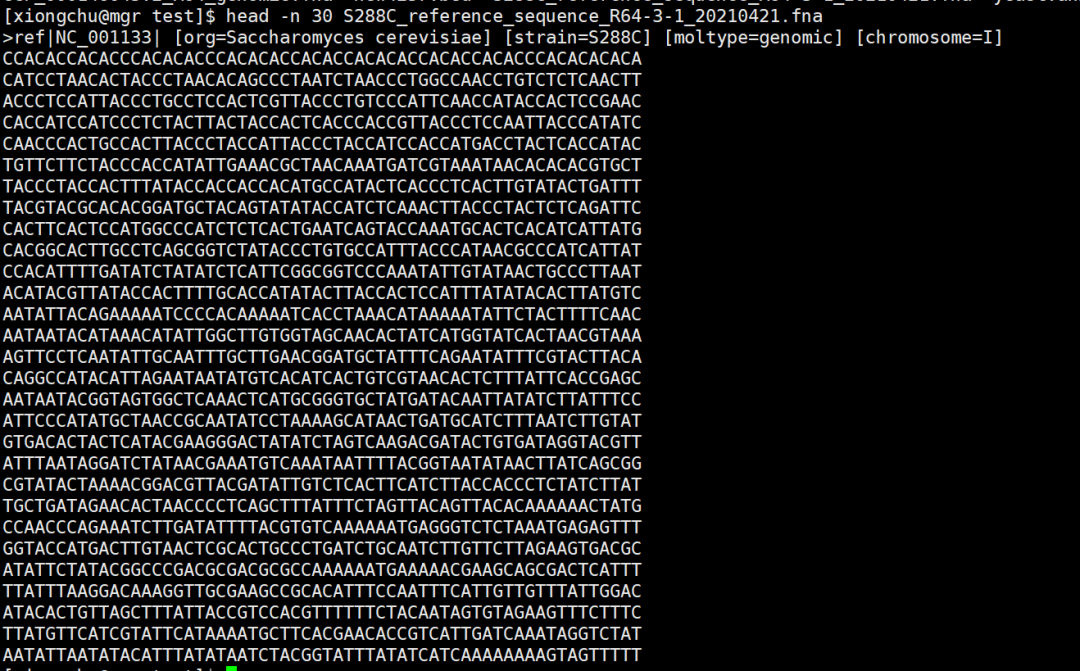
new4257-contigs.fa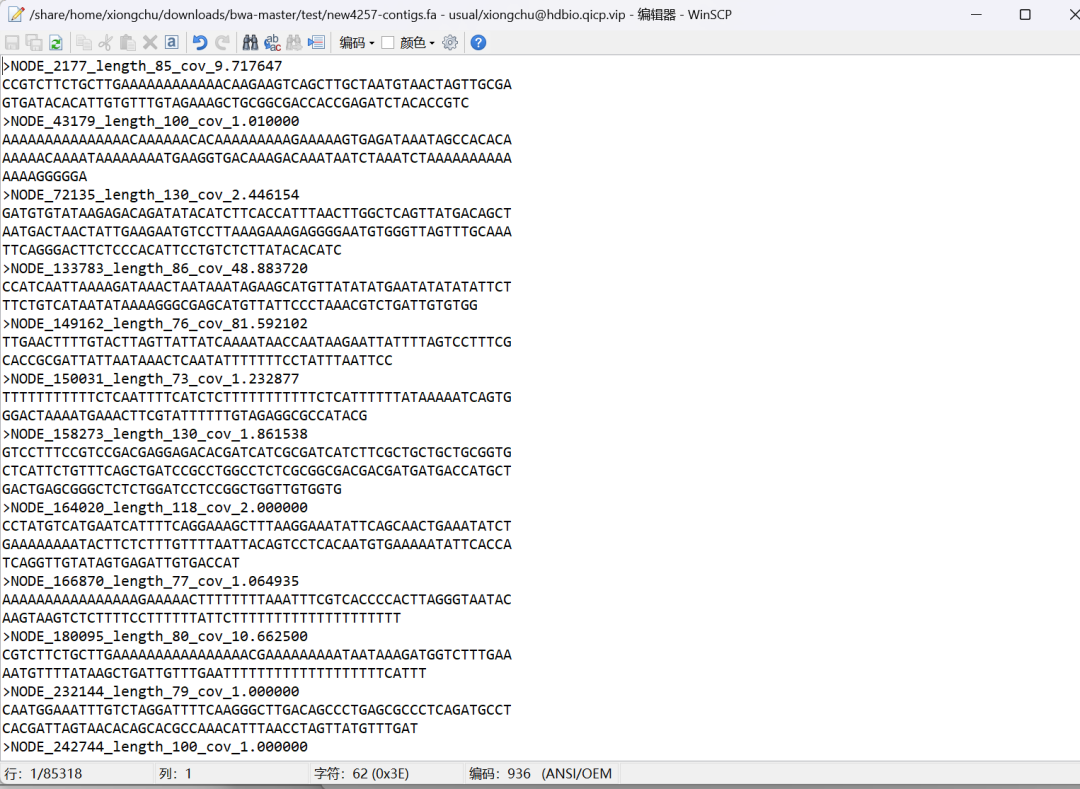
安装bwa
wget http://superb-sea2.dl.sourceforge.net/project/bio-bwa/bwa-0.7.12.tar.bz2tar jxf bwa-0.7.12.tar.bz2cd bwa-0.7.12makeecho 'PATH=$PATH:/your/bwa/path' >> ~/.bashrcsource ~/.bashrc
安装完成后,输入bwa 测试:
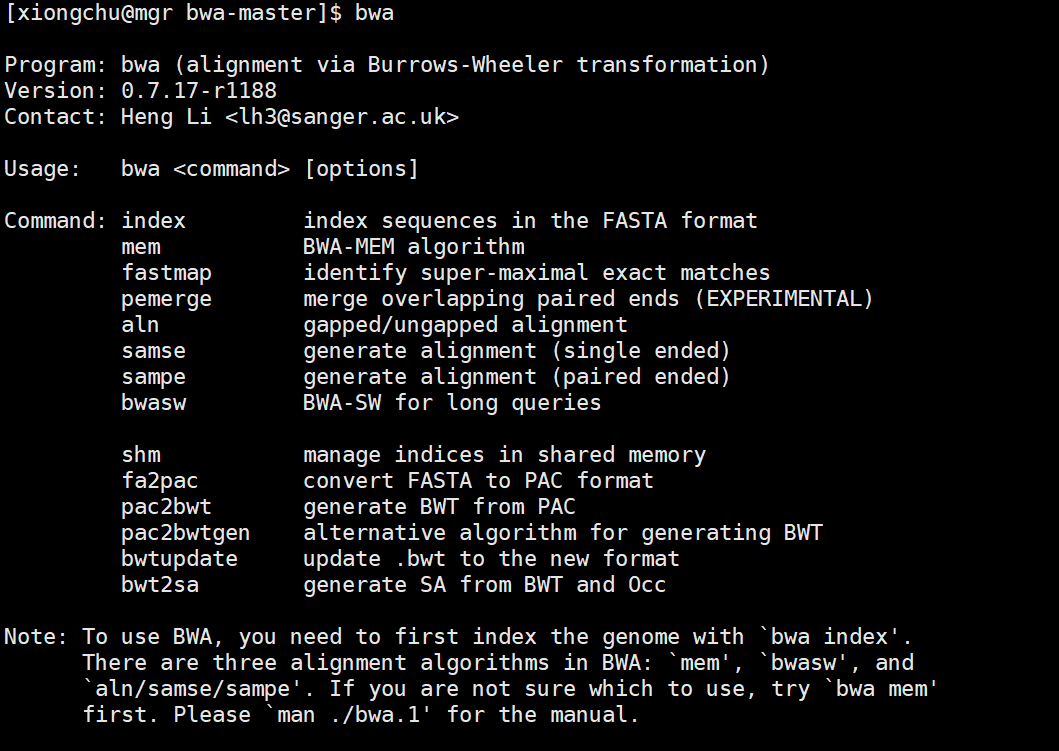
构建参考数据的索引
在进行序列比对之前,我们需要首先构建参考数据的索引。在这个教程中,我们将使用bwa工具来构建索引。在终端中执行以下命令:
bwa index -p yeast S288C_reference_sequence_R64-3-1_20210421.fna运行完成后,我们将得到以下文件:
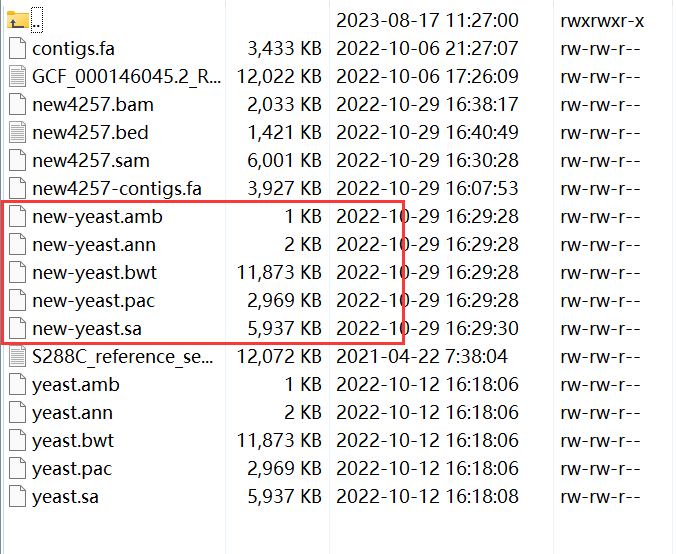
对待测序列进行比对
在此步骤中,我们将使用先前构建的索引将测序数据与参考序列进行比对。我们的测序数据文件为”new4257-contigs.fa”,并将比对结果输出到”new4257.sam”文件中。在终端中执行以下命令:
bwa mem -t 4 yeast new4257-contigs.fa > new4257.sam输出的文件格式如下:

转换sam文件格式为bam
为了后续对我们生成的比对信息处理,我们需要将文件转为bed文件,在转为bed文件之前,我们需要先转为bam文件
samtools是一个常用的处理SAM/BAM文件的工具。在这一步中,我们将使用samtools将SAM格式的比对结果转换为BAM格式。在终端中执行以下命令:
samtools view -@8 -b new4257.sam > new4257.bam对bam文件进行排序
在转为bed文件之前,我们先对BAM文件进行排序。这可以通过samtools的sort命令来实现。我们将使用”-O bam”参数指定输出格式为BAM,”-@ 10″参数指定使用10个线程进行排序,”-o”参数指定输出文件名为”new4257-sort.bam”。在终端中执行以下命令:
samtools sort -O bam -@ 10 -o new4257-sort.bam new4257.bam转换bam文件为bed文件
最后一步是将已排序的BAM文件转换为BED格式,以便进一步分析。我们将使用bedtools工具中的bamtobed命令来完成转换。在终端中执行以下命令:
bedtools bamtobed -i new4257-sort.bam > new4257.bed 我们一起来看下转换好的bed文件:
这样,我们就可以对我们比对后的结果进行进一步处理啦!
恭喜!你已经学会怎么使用bwa进行序列比对啦。现在,你可以使用生成的BED文件进行进一步的分析和注释。怎么样,是不是很简单!快和小果一起学习来吧!更多学习干货也要继续关注小果哦!
往期推荐
