bioawk一个超好用生信文本处理软件
今天小果给大家分享的是:bioawk,非常好用的工具,助力您更好更快地处理生信数据。
awk是linux系统中常用的处理文本文件的语言,它提供一个类编程环境来修改和重新组织文件中的数据。因为其功能强大,能够独当一面而广受使用者的好评。之所以叫 awk 是因为其取了三位创始人 Alfred Aho,Peter Weinberger, 和 Brian Kernighan 的 Family Name 的首字符。
在生信领域,数据格式复杂多样,对其数据文本的处理产生了新的挑战。awk虽然也能做,但是用起来终究还是没有那么得心应手。广大生信人就想,要是能有个专门处理生信数据文件的awk该有多好啊~
生信领域的大牛李恒,抓住了这一痛点,动手对awk的源码进行了改造。专门针对生信领域的文本处理工具——bioawk就由此应运而生了。而且是青出于蓝而胜于蓝。
在做生信的你还没用过的话,赶紧来跟小果上手吧~
软件安装
小果推荐用conda安装
conda install -y bioawk当然也可下载源码编译sudo apt-get install byacc git #安装gitgit clone git://github.com/lh3/bioawk.git #默认安装在当前目录git clone git://github.com/lh3/bioawk.git ${path}/bioawk #若加path则必须为空,因此可在想要安装的目录下起一个新名称,git会clone到其下cd bioawkmakeecho "export PATH=${path}/bioawk/:${PATH}" >> ~/.bashrc. ~/.bashrc

Bioawk的使用
一、帮助文档与使用方法
首先我们查看一下帮助文档,
#查看帮助文档bioawk -c help
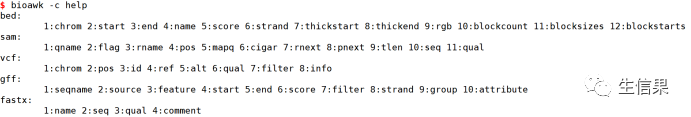
#bioawk的选项参数,使用方法bioawk
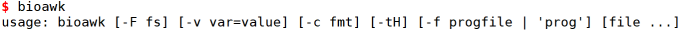
接下里是点图#点图
-F: 指定列与列之间的分隔符;-c: 支持的文件格式;-v: 设置自定义的变量-f: 从指定的文件中读取程序[file]: 最后指定的操作文件。
接下来小果会带大家由浅至深学习bioawk的使用。
二、处理fasta序列文件
(一)打印fasta序列ID、序列长度、GC含量
bioawk -cfastx '{print" ID:"$name" t length:"length($seq)" t GC:"gc($seq)" "}' xxx.fna-c fastx: 指定文件为fastx格式,包括fasta,fastq‘{code}’: 代码需要用{}括起来$name: 取文件中的变量name$seq: 取文件中的序列length(): length函数统计序列长度gc(): 序列中的gc含量XXX.fna: 指定的文件t: 两列信息用制表符t分割‘ ’: 脚本{}被视为单个文本字符串需要’’括起来,并且单引号表示输入的起始与结束。
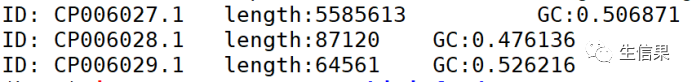
输入代码即可打印结果到终端。
(二)获取fasta序列的反向互补链序列
这是示例文件demo.fasta,的数据
#输出示例数据的5’->3’反向互补序列bioawk -c fastx '{ print ">" $name;print revcomp($seq) }' demo.fasta > revdemo.fasta使用revcomp()函数即可得到5’->3’反向互补序列revcomp($seq): 括号中输入需要处理的序列。> revdemo.fasta: 将结果输出到新的fasta文件 revdemo中

上述的单图是不是都很熟悉,都是平时我们做的比较多的图,我们创建完成后,就开始绘制组合图片把
这里使用ggpubr包中函数ggarrange()在一页上进行展示上述的结果
对ToothGrowth数据集的箱线图,点图组合展示:
ggarrange(Box_plot, Dot_plot,labels = c(“A”, “B”),ncol = 2, nrow = 1)
三、处理fastq文件
指定的文件格式依然选择 -c fastx,它能够自动识别序列是fastq 还是 fasta。Fastq文件比fasta文件多了测序质量的信息。
我们用于演示的fastq部分文件如下:
是不是就完成了呢,AB序号小伙伴可以自行调整
后面我们对#mtcars 数据集的条形图,散点图组合展示

(一)计算fastq序列的行数
构建测序数据分析 pipeline 时,当fastq 数据在做完 trimming 后,我们往往要关注剩下多少 reads,这是可以用 bioawk 进行快速统计。

bioawk -c fastx 'END {print NR}' SRR1039509_2.fastq
(二)统计特殊碱基开头的 reads 数
在一些特殊场合里,需要分析 reads ,用bioawk 可以很方便快速来完成。比如统计特殊碱基开头的 reads 数。
# 统计以GATTAC开头的reads的数量
bioawk -c fastx ‘$seq~/^GATTAC/ {++n} END { print n }’ my_fastq.tar.gz
# 统计以GATTAC开头的reads的数量bioawk -c fastx '$seq~/^GATTAC/ {++n} END { print n }' my_fastq.tar.gz
(三)查看GC含量大于60%的reads的质量情况
bioawk -c fastx '{if (gc($seq)>0.6) printf "%sn%snn", $seq, $qual}' my_fastq.tar.gz这里使用了if (gc($seq)>0.6) 条件判断语句,条件是gc含量大于60%。
打印printf,序列 $seq以及测序质量$qual
(四)计算序列平均质量分数
bioawk -c fastx ‘{print “>”$name; print meanqual($qual)}’ SRR1039509_2.fastq
我们可以运行bioawk计算每条序列的平均质量分数,通过函数meanqual()来实现。
返回序列的名称,以及质量分数。
(五)将fastq格式转换为fasta格式
bioawk -c fastx '{print ">"$name; print $seq}' SRR1039509_2.fastq
通过调用序列name变量并加上>,第二行打印seq变量,就将fastq文件转换为fastp文件了。
往期推荐

