十分钟GSEA从原理到绘图
GSEA原理
GSEA(Gene Set Enrichment Analysis)是一种用于分析基因表达数据的计算方法,旨在揭示基因集在特定生物学过程或疾病中的功能富集情况。它是一种广泛应用于生物信息学和系统生物学研究的工具。
GSEA的基本原理是通过将基因根据其表达模式进行排序,然后计算预定义的基因集在排序列表中的富集程度。这些基因集可以是与特定生物学过程、细胞信号通路、疾病相关的基因集合,或者是来自公共数据库(如基因集富集分析数据库)中的已知基因集。通过比较基因集在排序列表中的富集程度,可以确定哪些基因集与研究兴趣相关。GSEA的优点在于它不仅考虑单个基因的表达变化,而且关注整个基因集的富集情况。这种方法对于研究复杂的生物学过程和疾病机制非常有用,因为它能够捕捉到多个基因的协同调控和功能相关性。
小果有话要讲:在进行绘图的过程中,漂亮的图形是次要的,正确的分析才是重要的一环。
公众号回复“111”领取相关代码,代码编号:231001

GSEA实践
1.加载安装各种包
rm(list = ls())devtools::install_github("nicolash2/gggsea")BiocManager::install("GSVA")BiocManager::install("enrichplot")BiocManager::install("GSEABase")BiocManager::install("clusterProfiler")library(gggsea)library(ggplot2)library(GSVA)library(enrichplot)library('GSEABase')library(clusterProfiler)
2. 基因排序信息
#数据的准备至少包含两列为logFC和SYMBOL ID,根据logFC排序。deg<- read.csv("/Users/sansan/Desktop/软文/deg.csv",header = T,na.strings = T)#这里选择合适的方法read.delim()或者read.table()进行倒入数据。alldiff <- deg[order(deg$logFC,decreasing = T),]id <- alldiff$logFCnames(id) <- alldiff$gene_name#这里需要填入你存储genename的列
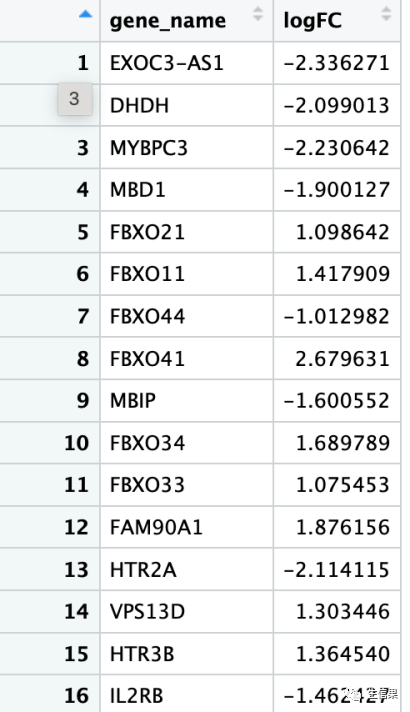
3.下载合适的数据集
一些已经预定义的功能基因集可以在http://www.gsea-msigdb.org/gsea/msigdb/index.jsp中下载,自定义基因集需要有较强的背景知识。
根据自己的需求进行下载~

点击进去后会出现下面的页面~
这里有两个版本,需要根据自己的需求进行选择性的下载,这里的genename为symbols形式,所以小果下载的为c2的symbols的格式的哦~

4. 导入分析
gmtfile <- "/Users/sansan/Downloads/c2.all.v2023.1.Hs.symbols.gmt.txt"hallmark <- read.gmt(gmtfile)gsea.re1<-clusterProfiler::GSEA(id,TERM2GENE=hallmark,verbose = T)#clusterProfiler::GSEA()这个函数可以?clusterProfiler::GSEA查看一下帮助,会获得更多的参数,例如可以调控的参数,详细的解释见附件#这一步是分析的主要步骤,分析结束后,会出现下面的字样

g1<-as.data.frame(gsea.re1)g1<-g1[order(g1$NES,decreasing = T),]
5. 出图
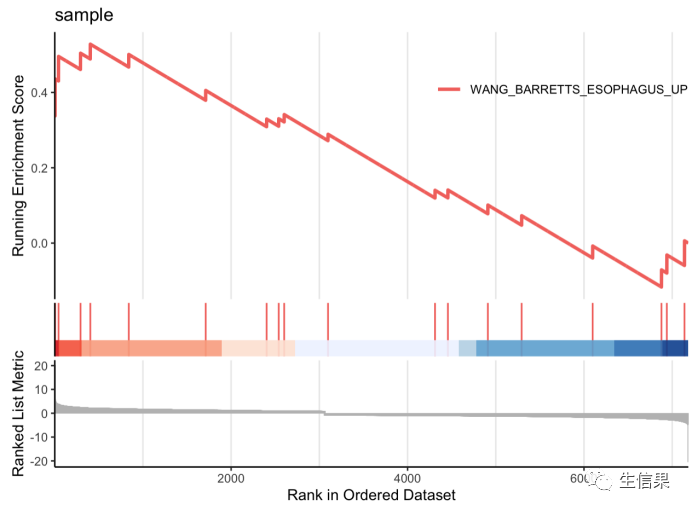
gseaplot2(gsea.re1,geneSetID = rownames(g1)[1],title = "sample",#标题base_size = 9.5,#大小尺寸rel_heights = c(1, 0.2, 0.4),#小图相对高度subplots = 1:3,#展示小图pvalue_table = FALSE,#p值表格ES_geom = "line"#line or dot)#输出单个的
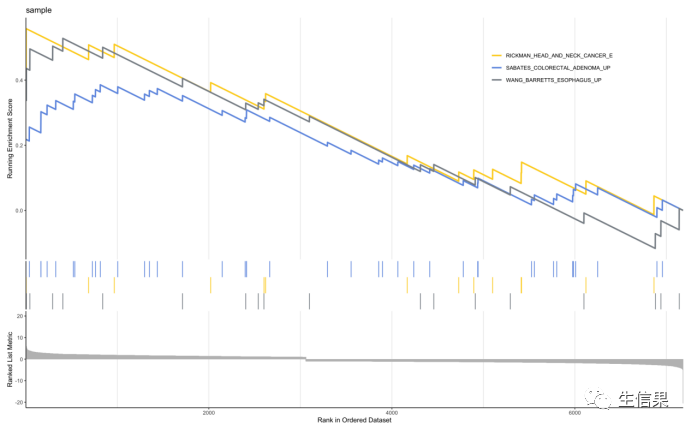
#展示多个基因集num=3gseaplot2(gsea.re1,geneSetID = rownames(g1)[1:num],title = "sample",#标题color = col_gsea1[1:num2],#颜色base_size = 9.5,#大小尺寸rel_heights = c(1, 0.2, 0.4),#小图相对高度subplots = 1:3,#展示小图pvalue_table = FALSE,#p值表格ES_geom = "line"#line or dot)
绘图函数的参数:gseaplot2(x,geneSetID,title = "",color = "green",base_size = 11,rel_heights = c(1.5, 0.5, 1),subplots = 1:3,pvalue_table = FALSE,ES_geom = "line")
附件:
在R中,clusterProfiler包中的GSEA()函数用于进行基因集富集分析。该函数提供了一些参数,可以用来调整筛选条件和扩大筛选范围。
下面是一些常用的参数和选项:
minGSSize:设置最小基因集大小。默认值为 10。你可以增加该值来筛选更大的基因集。
maxGSSize:设置最大基因集大小。默认值为 Inf(无穷大)。你可以减小该值来筛选更小的基因集。
pvalueCutoff:设置显著性水平的阈值。默认值为 0.05。你可以增大该值来筛选更显著的基因集。
fdrCutoff:设置FDR(False Discovery Rate)的阈值。默认值为 0.05。你可以增大该值来筛选更严格的基因集。
minGeneSetSize:设置最小基因集大小。默认值为 15。你可以增加该值来筛选更大的基因集。
maxGeneSetSize:设置最大基因集大小。默认值为 Inf(无穷大)。你可以减小该值来筛选更小的基因集。
这些参数可以根据你的需求进行调整,以扩大或缩小筛选条件。例如,如果你想要筛选更大的基因集或更显著的结果,可以增加minGSSize、pvalueCutoff或fdrCutoff的值。如果你想要筛选更小的基因集或更严格的结果,可以减小maxGSSize、pvalueCutoff或fdrCutoff的值。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~~或者也可以关注我们的官网也会持续更新的哦~
往期推荐

