果子带你揭示基因与临床数据预测生存预后的关键秘密
收录于话题
在生信分析的过程中,单因素、多因素COX回归分析常用来筛选与患者预后有关的因素,相信很多小伙伴对此并不陌生。
那么,如何在R中进行单因素、多因素COX回归分析?之前分享过绘制KM曲线,诺莫图展示COX结果,如何使用survminer包绘制Cox生存分析结果的森林图。
这篇文章都会给你答案,小果接下来就给大家进行超详细的代码实操展示。
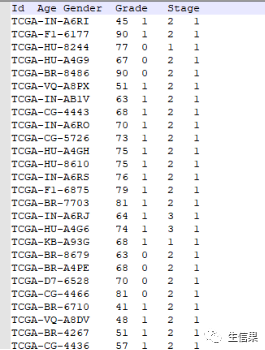
下面是示例样本的生存数据信息和临床数据文件

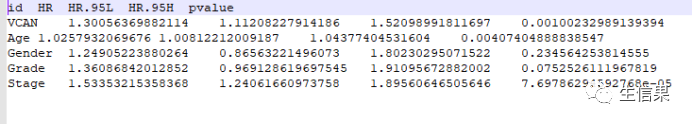
#install.packages('survival')library(survival) #引用包############定义森林图函数#############定义了一个名为bioForest的函数,#该函数有三个参数:coxFile、forestFile和forestCol,这些参数可以在函数被调用时传递实际值。bioForest=function(coxFile=null, forestFile=null, forestCol=null){#读取输入文件rt <- read.table(coxFile, header=T, sep="t", check.names=F, row.names=1)#用read.table函数从coxFile中读取数据,数据以Tab分隔,包含表头(header=T),#第一列作为行名(row.names=1),并将数据保存到名为rt的数据框中。gene <- rownames(rt)#将rt中的行名(基因名称)保存到gene变量中。hr <- sprintf("%.3f",rt$"HR")#将rt数据框中"HR"列的值保留三位小数并保存到hr变量中。hrLow <- sprintf("%.3f",rt$"HR.95L")hrHigh <- sprintf("%.3f",rt$"HR.95H")Hazard.ratio <- paste0(hr,"(",hrLow,"-",hrHigh,")")#使用paste0函数将"HR"、"HR.95L"和"HR.95H"列的值合并为一个字符串,#形成风险比和置信区间的表示,保存到Hazard.ratio变量中。pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))#用ifelse函数,如果rt数据框中的"pvalue"列小于0.001,#则将pVal变量设置为"<0.001",否则将其设置为保留三位小数的字符串表示。#输出图形pdf函数创建一个新的PDF绘图设备,指定输出文件名为forestFilepdf(file=forestFile, width=6.5, height=4.5)n <- nrow(rt)nRow <- n+1ylim <- c(1,nRow)layout(matrix(c(1,2),nc=2),width=c(3,2.5))#计算绘制图形所需的总行数,包括基因和每个基因对应的数据行。设置y轴的范围,从1到nRow。#使用layout函数设置绘图的布局,将绘制分为两列,第一列宽度为3英寸,第二列宽度为2.5英寸。#绘制森林图左边的临床信息xlim = c(0,3)par(mar=c(4,2.5,2,1))#用par函数设置图形的绘图参数。mar参数用于设置边缘空白的大小,包含4个值,依次为下、左、上、右的边缘空白大小。plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")text.cex=0.8text(0,n:1,gene,adj=0,cex=text.cex)text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex);text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)text(3.1,n:1,Hazard.ratio,adj=1,cex=text.cex);text(3.1,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1)#图中的坐标(1.5-0.50.2, n:1)处添加p-value的值,并通过adj=1参数将文本右对齐,cex=text.cex设置字体缩放。#然后在坐标(1.5-0.50.2, n+1)处添加'pvalue'文本,并设置字体加粗。#绘制右边的森林图par(mar=c(4,1,2,1),mgp=c(2,0.5,0))xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.05,col="darkblue",lwd=3)abline(v=1, col="black", lty=2, lwd=2)boxcolor = ifelse(as.numeric(hr) > 1, forestCol, forestCol)points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=2)axis(1)dev.off()}############定义森林图函数#############定义独立预后分析函数#定义了一个名为indep的函数,该函数有六个参数:expFile、cliFile、uniOutFile、multiOutFile、uniForest和multiForest,#这些参数可以在函数被调用时传递实际值。indep=function(expFile=null,cliFile=null,uniOutFile=null,multiOutFile=null,uniForest=null,multiForest=null){exp=read.table(expFile, header=T, sep="t", check.names=F, row.names=1) #读取表达文件cli=read.table(cliFile, header=T, sep="t", check.names=F, row.names=1) #读取临床文件#用read.table函数从expFile中读取数据,数据以Tab分隔,包含表头(header=T),#第一列作为行名(row.names=1),并将数据保存到名为exp的数据框中。#用read.table函数从cliFile中读取数据,数据以Tab分隔,包含表头(header=T),#第一列作为行名(row.names=1),并将数据保存到名为cli的数据框中。#数据合并sameSample=intersect(row.names(cli),row.names(exp))#获取两个数据框cli和exp行名(样本名称)的交集,并保存到sameSample变量中。exp=exp[sameSample,]cli=cli[sameSample,]rt=cbind(exp, cli)#单因素独立预后分析uniTab=data.frame()for(i in colnames(rt[,3:ncol(rt)])){cox <- coxph(Surv(futime, fustat) ~ rt[,i], data = rt)#使用coxph函数进行Cox比例风险模型分析,其中Surv(futime, fustat)表示生存时间和事件发生情况,#rt[,i]表示当前循环的列作为预测因子,data=rt表示使用rt数据框。coxSummary = summary(cox)uniTab=rbind(uniTab,cbind(id=i,HR=coxSummary$conf.int[,"exp(coef)"],HR.95L=coxSummary$conf.int[,"lower .95"],HR.95H=coxSummary$conf.int[,"upper .95"],pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))}write.table(uniTab,file=uniOutFile,sep="t",row.names=F,quote=F)bioForest(coxFile=uniOutFile, forestFile=uniForest, forestCol="green")
#多因素独立预后分析uniTab=uniTab[as.numeric(uniTab[,"pvalue"])<1,]rt1=rt[,c("futime", "fustat", as.vector(uniTab[,"id"]))]#从rt数据框中选择"futime"、"fustat"和uniTab数据框中的"pvalue"列对应的预测因子列,#并将它们合并为一个新的数据框rt1。multiCox=coxph(Surv(futime, fustat) ~ ., data = rt1)multiCoxSum=summary(multiCox)multiTab=data.frame()multiTab=cbind(HR=multiCoxSum$conf.int[,"exp(coef)"],HR.95L=multiCoxSum$conf.int[,"lower .95"],HR.95H=multiCoxSum$conf.int[,"upper .95"],pvalue=multiCoxSum$coefficients[,"Pr(>|z|)"])multiTab=cbind(id=row.names(multiTab),multiTab)write.table(multiTab,file=multiOutFile,sep="t",row.names=F,quote=F)bioForest(coxFile=multiOutFile, forestFile=multiForest, forestCol="red")}
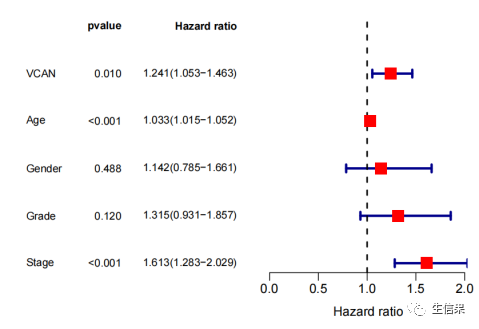
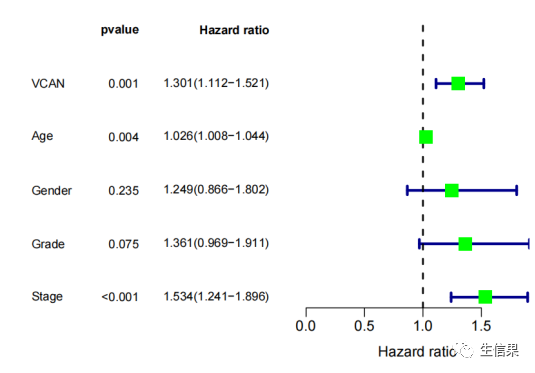
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~或者也可以关注我们的官网也会持续更新的哦~
往期推荐

点击“阅读原文”立刻拥有
↓↓↓