用R语言包GSVA,将数据转化为样本与基因关联矩阵,帮助你更直观且高效的分析
点击蓝字 关注我们
install.packages("GSVA") #安装GSVA语言包library(GSVA) #加载语言包
# 导入所需包> install.packages("GSVA")> install.packages("limma")> library(GSVA)> library(limma)# 导入示例基因表达数据,这里假设有一个基因表达矩阵data_matrix,行表示基因,列表示样本# 假设还有一个基因集列表gene_sets_list,包含若干个基因集,每个基因集是一个字符向量,表示基因的Symbol或Entrez ID# 假设还有一个对应样本的类别标签group_labels,用于区分不同生物学状态或条件# 生成示例数据,这里使用R的内置数据集iris> data_matrix <- as.matrix(iris[, 1:4])> gene_sets_list <- list(+ upregulated_genes = c("geneA", "geneB", "geneC"),+ downregulated_genes = c("geneX", "geneY", "geneZ")+)> group_labels <- factor(iris$Species)# 运行GSVA算法> gsva_result <- gsva(data_matrix, gene_sets_list, method = "gsva", mx.diff = FALSE)# 可视化结果# 可以使用heatmap展示基因集在样本中的表达模式> heatmap(gsva_result$es.obs, Rowv = NA, Colv = NA, col = colorRampPalette(c("blue", "white", "red"))(100))
# 使用boxplot或violinplot展示不同基因集在不同生物学条件下的得分分布> boxplot(gsva_result$es.obs ~ group_labels, col = rainbow(nlevels(group_labels)))
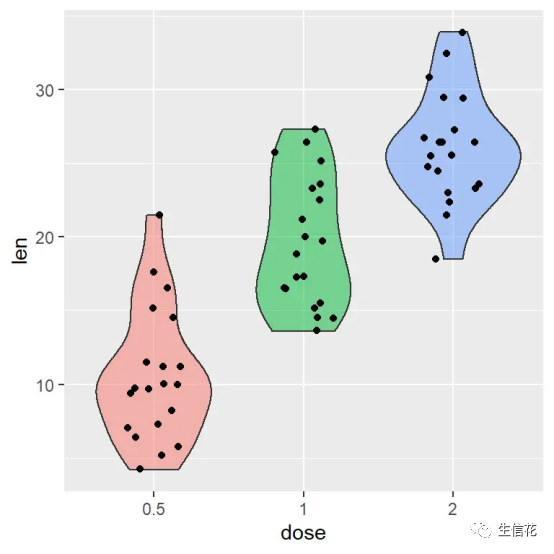
# 或者使用violinplot> vioplot(gsva_result$es.obs ~ group_labels, col = rainbow(nlevels(group_labels)))
# 使用PCA或t-SNE等降维方法,将样本投影到低维空间,并根据GSVA得分进行着色> pca_result <- prcomp(t(gsva_result$es.obs))> plot(pca_result$x[, 1], pca_result$x[, 2], col = group_labels)
以上就是对R语言包GSVA的简单介绍啦,GSVA作为一种无参数的基因集分析工具,在生物学研究中发挥着重要作用。通过将基因表达数据转换为样本和基因集之间的关联矩阵,GSVA为生物学家提供了一种直观、高效的分析手段,有助于揭示基因集在不同生物学过程中的潜在功能和生物学意义。未来随着技术的不断发展,GSVA将继续在生物学研究中发挥重要作用,并为我们深入理解基因表达调控和生物学过程的奥秘提供有力支持。
小花有话说
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小花哦~~或者也可以关注我们的官网也会持续更新的哦~ http://www.biocloudservice.com/home.html



长按识别二维码关注我们哟


(点击阅读原文跳转)
![]() 点一下阅读原文了解更多资讯
点一下阅读原文了解更多资讯
