蛋白质组学的瑞士军刀:mpwR让实验分析变得如此简单!

大家好,今天小果要给大家介绍的是一个2023年1月1日发表在Bioinformatics期刊上名为mpwR的R包,它的主要目的是对基于质谱(MS)的蛋白质组学无标签工作流程进行标准化比较。无论你的数据依赖性或非数据依赖性的光谱获取方式,mpwR都可以帮助你轻松比较样本准备程序、液相色谱(LC)-MS设置的组合以及软件内和软件间的关键性能指标的差异。这款用户友好的设计使得我们可以方便地比较各种分析中这些因素的影响,而且可以对比无限数量的分析结果。此外,mpwR还支持Bottom-up蛋白质组学常用的软件的输出,如ProteomeDiscoverer、Spectronautil、MaxQuant和DIA-NN等。
公众号回复111领取代码,代码编号:231108
下面小果就要拔出这把宝刀,看看mpwR的锋芒!我将带领大家了解如何使用mpwR进行数据导入和下游分析。我们将会重点介绍如何标记识别数量、数据完整性、定量和保留时间精度等等。
安装
devtools::install_github("OKdll/mpwR", dependencies =TRUE)
加载r包和导入数据
首先,我们需要加载一些必要的R包:
library(mpwR)library(flowTraceR)library(magrittr)library(dplyr)library(tidyr)library(stringr)library(tibble)library(ggplot2)library(flextable)
接下来,我们需要导入数据。可以使用prepare_mpwR函数来完成这个任务。请将所有输出文件放在一个文件夹中,并按照命名指南对文件进行命名。不允许使用其他文件或子文件夹。
files <-prepare_mpwR(path ="Path_to_Folder_with_files")为了让大家更好地理解工作流程,小果就直接用示例数据来演示。我们可以使用create_example函数来创建示例文件:
files <-create_example()通过以上步骤,我们就可以使用mpwR完成数据导入了。
Identifications数量如何确定识别数量?可以使用get_ID_Report函数来完成这个任务。

ID_Reports <-get_ID_Report(input_list = files)这个函数会为每个分析生成一个ID Report,并将其存储在一个列表中。现在,让我们来看一下如何轻松访问报告中的数据。假设我们要查看DIA-NN的报告:
flextable::flextable(ID_Reports[["DIA-NN"]])
接着我们看看如何生成条形图。可以使用plot_ID_barplot函数来完成这个任务。
ID_Barplots <-plot_ID_barplot(input_list = ID_Reports, level ="ProteinGroup.IDs")这个函数会根据输入的报告列表(ID_Reports)和层级(”ProteinGroup.IDs”)生成条形图,并将它们存储在一个列表中。现在,让我们来看一下如何轻松访问单个条形图。假设我们要查看DIA-NN的条形图:
ID_Barplots[["DIA-NN"]]
我们还可以画出箱线图进行总结。
plot_ID_boxplot(input_list = ID_Reports, level ="ProteinGroup.IDs")
数据完整性
报告
首先,让我们来了解一下如何生成数据完整性报告。可以使用get_DC_Report函数来完成这个任务。
DC_Reports <-get_DC_Report(input_list = files, metric ="absolute")DC_Reports_perc <-get_DC_Report(input_list = files, metric ="percentage")
这个函数会根据输入的文件列表(files)和指标(metric)生成数据完整性报告,并将它们存储在一个列表中。现在,让我们来看一下如何轻松访问单个数据完整性报告。假设我们要查看DIA-NN的报告:
flextable::flextable(DC_Reports[["DIA-NN"]]
可视化
除了查看报告,我们还可以使用mpwR生成各种可视化图表来展示数据分析的结果。可以使用plot_DC_barplot函数来完成这个任务。
DC_Barplots <-plot_DC_barplot(input_list = DC_Reports, level ="ProteinGroup.IDs", label ="absolute")这个函数会根据输入的数据完整性报告列表(DC_Reports)和层级(”ProteinGroup.IDs”)生成条形图,并以可视化的方式展示数据分析的结果。
可以通过下面的代码画出个体的柱状图。
DC_Barplots[["DIA-NN"]]
同样地,我们可以使用plot_DC_barplot函数生成百分比形式的条形图:
DC_Barplots <-plot_DC_barplot(input_list = DC_Reports_perc, level ="ProteinGroup.IDs", label ="percentage")[["DIA-NN"]]这个命令会根据输入的数据完整性报告列表(DC_Reports_perc)和层级(”ProteinGroup.IDs”)生成百分比形式的条形图,并只显示DIA-NN的结果。

汇总
汇总信息可以通过plot_DC_stacked_barplot生成堆叠柱状图。
绝对值
plot_DC_stacked_barplot(input_list = DC_Reports, level ="ProteinGroup.IDs", label ="absolute")
百分比
plot_DC_stacked_barplot(input_list = DC_Reports_perc, level ="ProteinGroup.IDs", label ="percentage")
Missed Cleavages
报告
我们可以用get_MC_Report 函数查看Missed Cleavages的绝对值或者百分比。
MC_Reports <-get_MC_Report(input_list = files, metric ="absolute")MC_Reports_perc <-get_MC_Report(input_list = files, metric ="percentage")
结果会被存储在list中,我们可以直接根据索引查看。
flextable::flextable(MC_Reports[["Spectronaut"]])
可视化
个体
绝对值
每个MC报告可以用plot_MC_barplot从前体到蛋白组水平绘制。生成的条形图存储在一个列表中。
MC_Barplots <-plot_MC_barplot(input_list = MC_Reports, label ="absolute")接着也是通过索引来访问。
MC_Barplots[["Spectronaut"]]
百分比
plot_MC_barplot(input_list = MC_Reports_perc, label ="percentage")[["Spectronaut"]]
总结
同样是通过堆叠条形图的方法来呈现。
绝对值
plot_MC_stacked_barplot(input_list = MC_Reports, label ="absolute")
百分比
plot_MC_stacked_barplot(input_list = MC_Reports_perc, label ="percentage")
保持时间精度
准备
变异系数(CV)可以通过get_CV_RT计算。
CV_RT <-get_CV_RT(input_list = files)可视化
plot_CV_density(input_list = CV_RT, cv_col ="RT")
定量精度
肽水平
准备
CV_LFQ_Pep <-get_CV_LFQ_pep(input_list = files)可视化
plot_CV_density(input_list = CV_LFQ_Pep, cv_col ="Pep_quant")
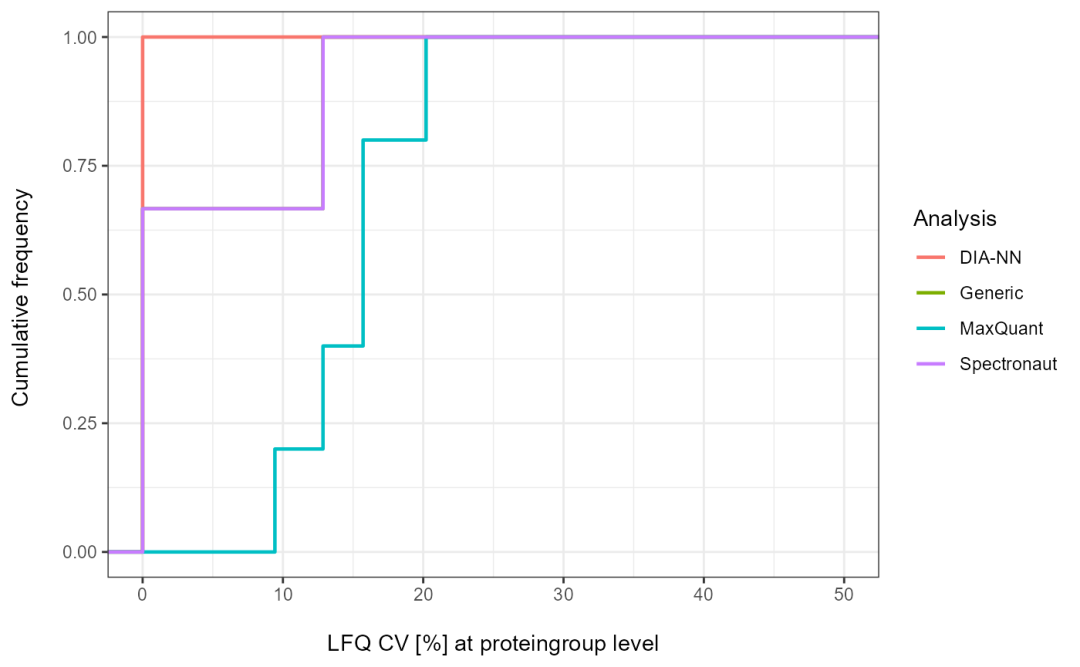
Proteingroup-level准备
CV_LFQ_PG <-get_CV_LFQ_pg(input_list = files)可视化
plot_CV_density(input_list = CV_LFQ_PG, cv_col ="PG_quant")
Upset Plot准备
Upset_prepared <-get_Upset_list(input_list = files, level ="ProteinGroup.IDs")
可视化
plot_Upset(input_list = Upset_prepared, label ="ProteinGroup.IDs")

总结
mpwR提供了一个系统的方法来比较蛋白质组工作流程,并使研究人员能够获取有关鉴定、数据完整性、定量精确性和其他性能指标。大家之后进行蛋白质组相关的研究时不要忘记了mpwR这把瑞士军刀哦!
参考文献
[1] Kardell O, Breimann S, Hauck SM. mpwR: an R package for comparing performance of mass spectrometry-based proteomic workflows. Bioinformatics. 2023 Jun 1;39(6):btad358. doi: 10.1093/bioinformatics/btad358. PMID: 37267150; PMCID: PMC10265443.
[2] https://okdll.github.io/mpwR/articles/Workflow.html


往期推荐