Qualimap:一款强大的生信绘图工具,助力你的测序数据分析

大家好,小果今天和大家分享一款生信绘图工具——Qualimap。Qualimap是一款非常实用的生物信息学软件,它可以帮助你对基因组、转录组和表观遗传组的测序数据进行质量评估和分析。
Qualimap的功能主要包括:对测序数据的BAM文件进行覆盖度、重复度、插入片段长度等进行统计和可视化;对基因组变异、结构变异、表观遗传标记等VCF文件进行检测和注释;对RNA-seq数据进行基因表达量计算、差异表达分析、功能富集分析等;对ChIP-seq数据进行质量控制,检查峰值的富集度、分布、重叠等指标;对多样本的数据进行比较分析,绘制PCA图、热图、箱线图等;对基因组注释文件进行解析,提取基因组特征信息。
跟着小果一起测试一下吧!
官方下载网址:http://qualimap.conesalab.org/
或者GitHub上的源代码https://github.com/EagleGenomics-cookbooks/QualiMap
解压并安装R包:
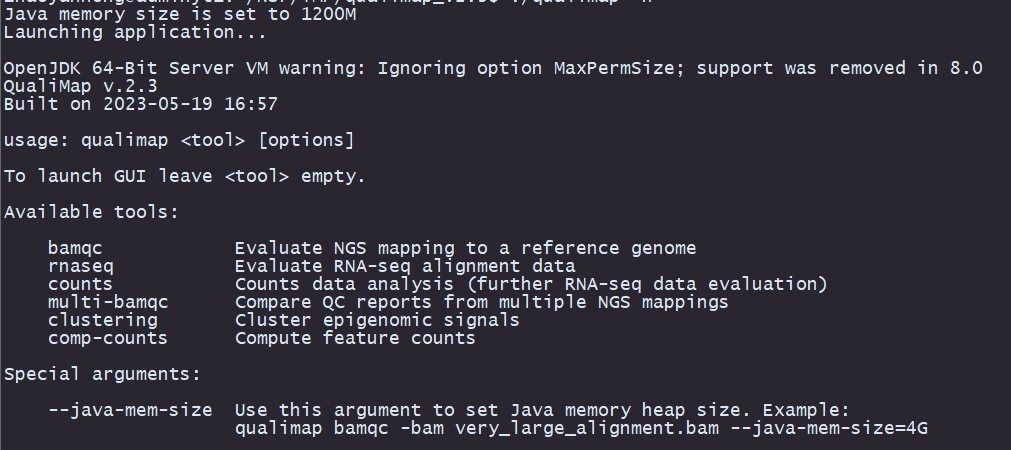
unzip qualimap_v2.3.zipcd qualimap_v2.3/Qualimap需要安装版本6或更高版本的Java和3.1或更高版本的R环境,Qualimap需要以下R包:optparse(可从CRAN获得),NOISeq,Repitools,Rsamtools,GenomicFeatures,rtracklayer(可从Bioconductor获得)。您可以手动安装这些包,也可以通过执行安装文件夹中找到的脚本来安装它们:sudo Rscript $QUALIMAP_HOME/scripts/installDependencies.r来看一下qualimap的用法和功能:·bamqc:评估NGS测序数据与参考基因组的比对情况。·rnaseq:评估RNA-seq测序数据的比对情况。·counts:对计数数据进行分析(进一步评估RNA-seq数据)。·multi-bamqc:比较多个NGS测序数据的质量控制报告。·clustering:聚类表观遗传信号。·comp-counts:计算特征计数。
下面我们来做一个使用Qualimap进行BAM质量控制分析的示例:
# 运行BAM QC分析qualimap bamqc -bam example.bam -outdir output_dir -outformat PDF:HTML# 运行BAM QC分析并指定参考基因组qualimap bamqc -bam example.bam -outdir output_dir -outformat PDF:HTML -gff example.gff出现了错误/(ㄒoㄒ)/~~
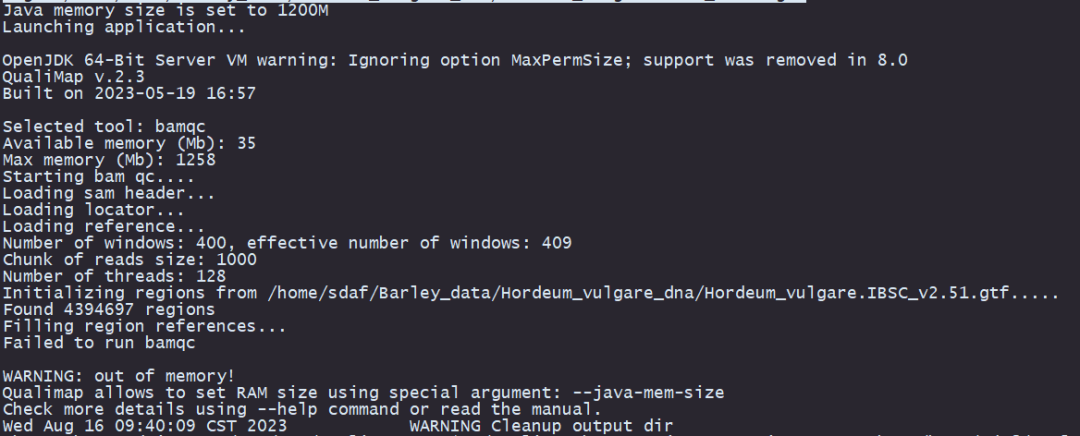
错误信息显示因为内存不足,Java内存大小被设置为1200M,在运行过程中出现了内存不足的警告。那么增大内存布局好了吗?果然不出小果所料,增加–java-mem-size=4G后再次运行,这个问题解决了 o(* ̄▽ ̄*)ブ,但是错误又来了。
qualimap bamqc -bam example.bam --java-mem-size=4G -outdir output_dir -outformat PDF:HTML -gff example.gff错误信息显示在计算报告时,程序出现了一个异常:无法连接到X11窗口服务器。这个问题可能是由于Java虚拟机使用DISPLAY环境变量来检测X11系统是否可用,但有时这个变量被操作系统或某些应用程序错误地设置了。
要解决这个问题,可以尝试取消设置DISPLAY变量,方法是在命令行中输入unset DISPLAY或export DISPLAY=:0。此外,你还可以使用Java的一个特殊选项-Djava.awt.headless=true来禁用显示要求。

要启用这个选项,你可以在系统中设置JAVA_OPTS变量,或者在qualimap脚本中修改java_options变量,例如:
java_options="-Djava.awt.headless=true -Xmx$JAVA_MEM_SIZE -XX:MaxPermSize=1024m"这里我选择了修改java_options变量,再次运行上面的命令,这次成功啦!生成了一个结果文件夹output_dir,里面包含了3给文件和3给文件夹,其中我们主要看qualimapReport.html网页文件。
qualimapReport.html网页文件中包含表格形式的统计信息和图片信息。
表格信息包括:
全局部分包含有关读数总数、映射读数数、成对端映射性能、读长分布、剪切读数数和重复率(根据读对齐的起始位置估算)的信息。
ACGT内容部分报告映射读数中的核苷酸含量和GC百分比。
覆盖部分提供覆盖深度的平均值和标准差的信息。
映射质量部分报告映射读数的平均映射质量。
插入大小部分提供有关插入大小分布的平均值、标准差和百分位数的信息(如果适用),基于SAM文件的TLEN字段。
错配和插入/缺失部分报告一般对齐错误率、错配总数和插入/缺失总数,从CIGAR值计算。此外,还提供了同聚物插入/缺失在总插入/缺失中的比例。请注意,错误率和错配度量基于SAM记录的可选字段(NM用于编辑距离,MD用于错配),如果这些字段在SAM文件中缺失,则不报告这些特征。染色体统计部分提供有关每个染色体(由SAM文件头定义)的映射碱基数、覆盖深度的平均值和标准差的信息。对于基于区域的分析,区域内提供额外信息,例如正确链读数的数量。
图片信息包括:
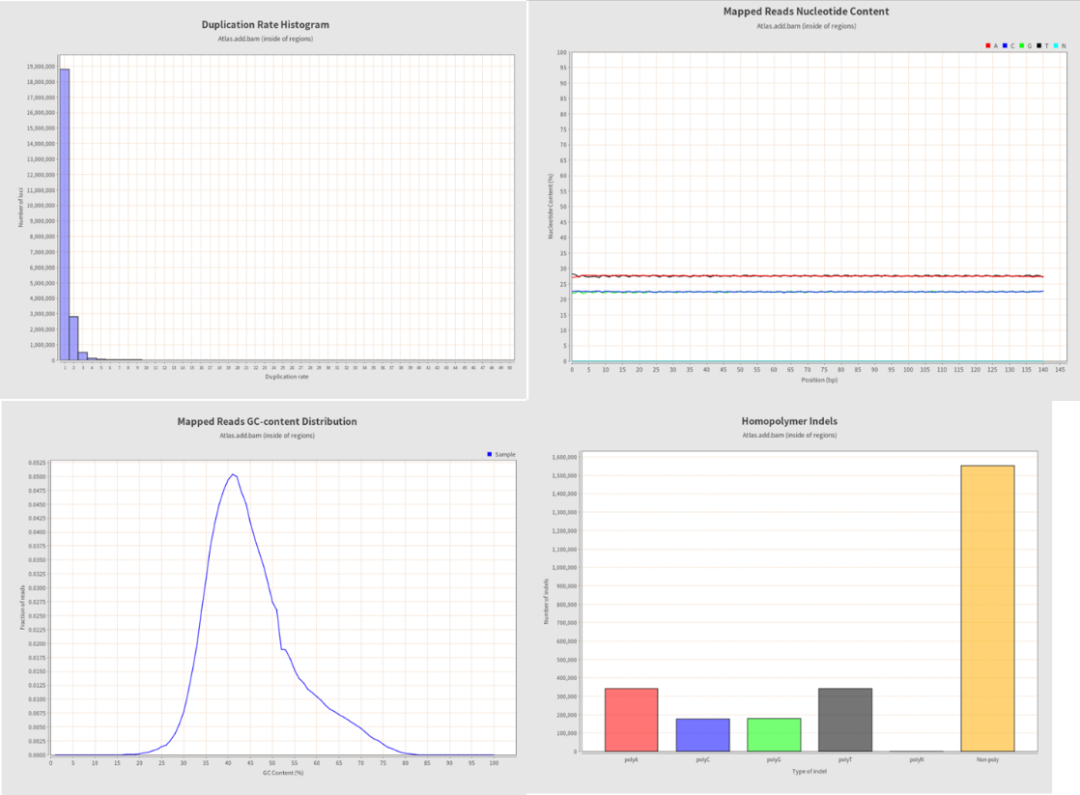
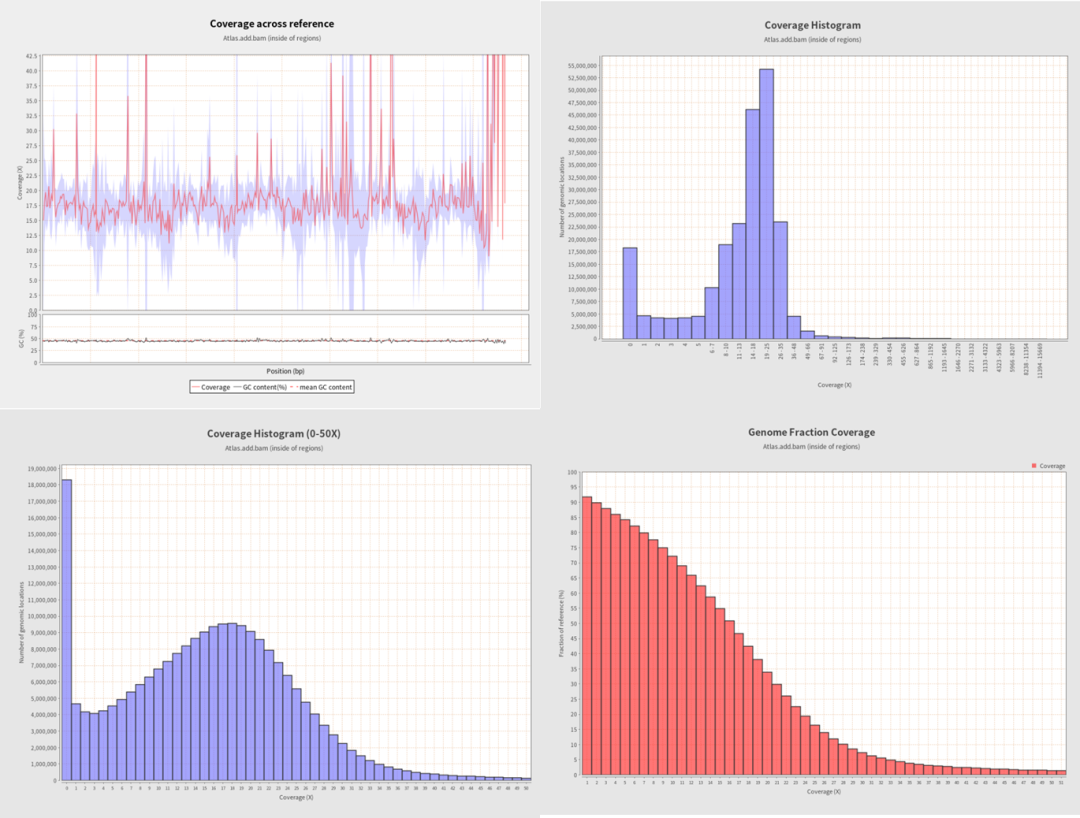
覆盖率分布图。由两个图形组成,上图显示了参考序列的覆盖率分布(红线)和覆盖率偏差。下图显示了参考序列的GC含量(黑线)及其平均值(红色虚线)。
覆盖率直方图显示具有给定覆盖率的基因组位置数。x轴的箱通过聚合一些覆盖值来方便地缩放,以便在存在常见NGS覆盖峰值的情况下也能产生具有代表性的直方图。
覆盖率直方图(0-50X)显示具有给定覆盖率的基因组位置数。在这张图中,覆盖率大于50X的基因组位置被分组到最后一个箱中。这样做可以获得对覆盖率最常见值的更高分辨率。
基因组分覆盖图。了解至少具有给定覆盖率的参考序列被测序了多少。这张图应该按照以下示例进行解释:
如果目标是至少25X的覆盖率(x轴),那么y轴应该是约83%。
重复率直方图。显示重复读取起始位置的分布。
映射reads上的核苷酸含量贴图。此图显示映射到reads每个位置的核苷酸含量。
GC含量分布图。此图显示每个映射读数的GC含量分布。如果与预先计算的基因组分布进行比较,此图可以检查是否存在GC含量偏移。
如果你想了解更多的信息,可以参考qualimap文档,网址是:http://qualimap.conesalab.org/doc_html/index.html# 。
参考文献:Okonechnikov, K., Conesa, A., & García-Alcalde, F. (2015). “Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data.” Bioinformatics, btv566
往期推荐
1.搭建生信分析流水线,如工厂一样24小时运转Snakemake——进阶命令 2.比blast还优秀的序列比对工具?HMMER来了 3.对单细胞分析毫无头绪?让popsicleR领你入门 4.小果带你绘制ROC曲线评估生存预测能力 5.软件包安装、打怪快又好,1024G存储的生信服务器;还有比这更省钱的嘛!!!