怎样才能在信息爆炸的时代中快速获取准确的生信数据呢?Rentrez包,NCBI数据库的超级通行证!


公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240402
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager ")BiocManager::install("rentrez") # 在BiocManager环境下安装rentrez查看是否安装成功packageVersion("rentrez") # 查看rentrez版本

library(rentrez) # 载入rentrez包entrez_dbs() # 查找可用数据库的列表

entrez_db_summary("nuccore") # 查看核酸数据库信息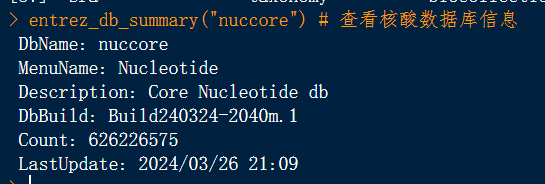
r_search <- entrez_search(db="pubmed", term="R Language") # 在pubmed数据库中检索R语言相关信息该命令的返回值是一个列表,我们可以直接查看它的信息。r_search # 打印r_search内容

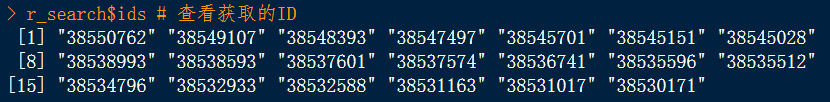
pubmed_id <- "12345678" # 替换为你的文献IDarticle <- entrez_fetch(db="pubmed", id=pubmed_id, rettype="abstract")# 使用entrez_fetch函数下载文献信息print(article) # 打印下载的文献信息

accession_number <- "NC_000913"sequence <- entrez_fetch(db = "nuccore", id = accession_number, rettype = "fasta", retmode = "text")out_file <- "Ecoli.fasta"write(sequence, file = out_file, append = TRUE)

小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!


定制生信分析
服务器租赁
扫码咨询小果

往期回顾
|
01 |
|
02 |
|
03 |
|
04 |