小白也能轻松上手!基因组数据处理新思路,GenomicRangesR包助你深入剖析基因组数据!

大家好,我是小果!基因组数据处理让你头疼不已?海量数据让你迷失方向?别担心,GenomicRanges包来拯救你!这款强大的R语言工具,专为基因组学设计,从读取到分析,一站式搞定你的数据处理需求。接下来,小果将带你轻松上手GenomicRanges包,解锁基因组数据的无限可能!快来一起探索吧!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
lGenomicRangesR包简要说明
GenomicRangesR包是R语言生物信息学分析领域中的一个重要工具,专为处理和分析基因组学数据而设计。这个包提供了基础的数据结构,使得我们能够轻松地对基因组中的基因、转录本、染色体等生物学特征的位置和范围信息进行操作和管理。
GenomicRangesR包的核心是其数据结构,特别是GRanges对象。这个对象能够表示基因组范围的集合,每个范围都有明确的起点和终点位置,从而精确地描述基因组上的各种特征。此外,GRanges还支持附加元数据,如链方向、score值和GC含量等,进一步丰富了其对基因组特征的描述能力。
在功能方面,GenomicRangesR包提供了丰富的函数和方法,用于对基因组范围进行各种操作,如合并、分割、平移、过滤和排序等。此外,GenomicRangesR包还具有良好的可视化能力。它可以与其他数据可视化包(如ggplot2、Gviz等)无缝结合,帮助我们绘制基因组范围的图形和图表,直观地展示基因的位置、分布和特征。
lGenomicRangesR包代码实操
小果这里将用一段代码和小伙伴们一起学习GenomicRangesR包进行基因组数据分析的一个典型流程。本次介绍的R包操作会占用内存较大,小果建议使用服务器,欢迎小伙伴们联系小果租赁性价比居高的服务器!
公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240520-1
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

Ø加载GenomicRanges包:首先检查是否已安装,未安装则进行安装并加载。
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("GenomicRanges")library(GenomicRanges)
Ø创建基因组区间对象:小果此处使用IRanges函数创建基因组区间,并用GRanges添加了相关的序列信息。然后创建了一个名为meta的DataFrame对象,其中包含了要添加的元数据(基因名称和表达水平)并将其绑定到gr对象的元数据列(mcols)。再使用elementMetadata函数将这个元数据对象与gr对象关联起来,从而为每个区间添加了元数据。
gr <- GRanges(seqnames = c("chr1", "chr2", "chr3"), ranges = IRanges(start = c(100, 200, 300), end = c(150, 250, 350)), strand = c("+", "-", "+"))meta <- DataFrame(gene_name = c("Gene1", "Gene2", "Gene3"), expression_level = c(10.5, 20.1, 15.3))gr$mcols <- metaprint(gr)gr_positive_strand <- gr[strand(gr) == "+"]

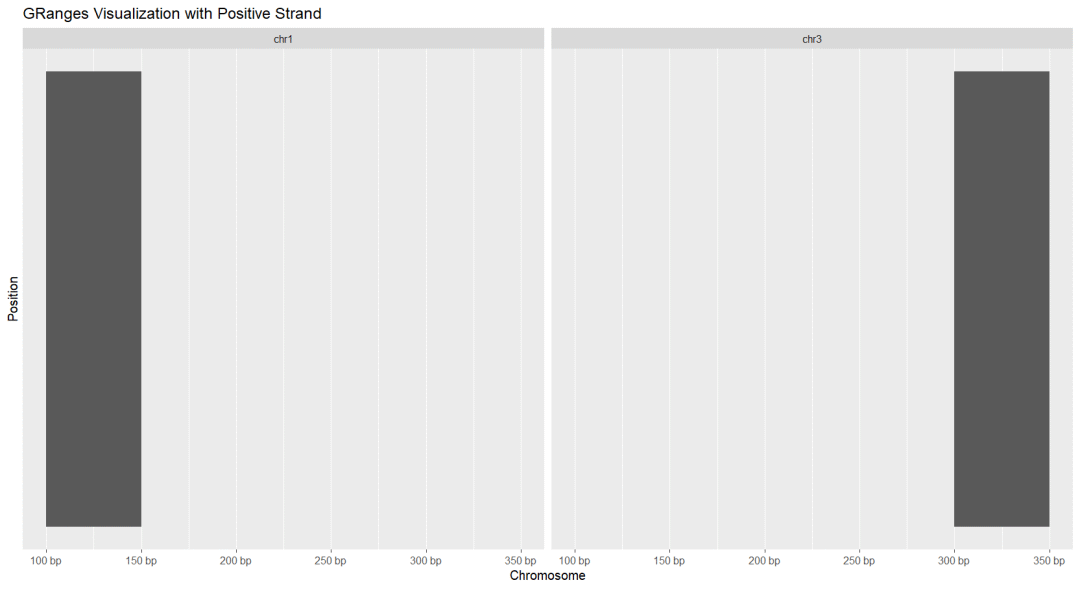
Ø将GRanges对象可视化:使用ggbio的autoplot函数来可视化GRanges对象所有区间,不论正负链(省略which参数)
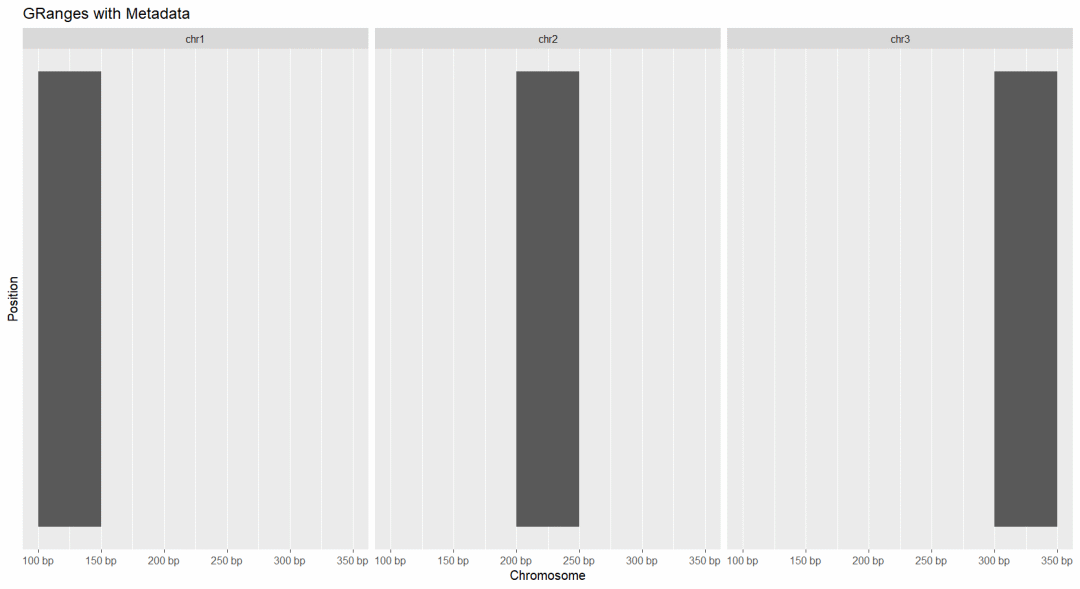
autoplot(gr_positive_strand) +labs(title = "GRanges Visualization with Positive Strand", x = "Chromosome", y = "Position")autoplot(gr) +labs(title = "GRanges with Metadata", x = "Chromosome", y = "Position")
Ø区间查询与操作:使用findOverlaps等函数查询区间间的重叠,并进行合并、交集操作以及区间的统计摘要。然后小果创建了一个查询对象query_gr,用于查询染色体chr1上的区间,并找出与gr对象重叠的部分。
gr2 <- GRanges("chr1", IRanges(start = 120, end = 220), "+")overlaps <- findOverlaps(gr, gr2)intersection <- gr[queryHits(overlaps)]print(intersection)cat("Number of intervals:", length(gr), "n")cat("Average interval length:", mean(gr_lengths), "n")query_gr <- GRanges("chr1", IRanges(start = 1, end = 500), "+")query_result <- findOverlaps(query_gr, gr)
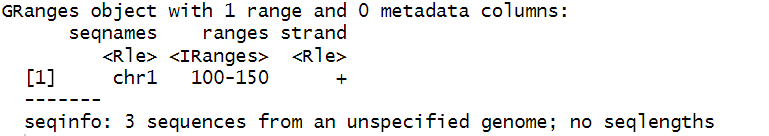

Ø数据可视化:利用GenomicRanges的绘图功能,直观展示基因组区间。小果先将查询结果转换为数据框overlap_df,计算每个区间的长度,并绘制区间长度的直方图。一个条形图展示基因表达水平与区间宽度的关系,一个点图展示区间宽度与基因表达水平的关系。最后,使用heatmap函数绘制了一个基因表达水平的热力图。
overlap_df <- as.data.frame(query_result)gr_lengths <- width(gr)hist(gr_lengths, breaks=20, col="pink", main="Histogram of Interval Lengths", xlab="Length")df <- as.data.frame(gr)ggplot(data = df, aes(x = seqnames, y = width, fill = "gene_name")) +geom_bar(stat = "identity") +theme_minimal() +labs(title = "Gene Expression Levels", x = "Chromosome", y = "Interval Width")ggplot(data = df, aes(x = width, y = mcols.expression_level)) +geom_point() +theme_minimal() +labs(title = "Gene Expression Levels vs Interval Width", x = "Interval Width", y = "Expression Level")heatmap(data.matrix(df), Rowv = NA, Colv = NA,main = "Gene Expression Heatmap")

l文章小结
本期小果为小伙伴们介绍了一个功能强大、易于使用的工具——GenomicRangesR包以及它处理基因组学数据的流程。我们先创建了一个GRanges对象gr,表示基因组中的基因区间,后又创立了一个元数据对象meta,将元数据添加到GRanges对象中,并使用了ggbio的autoplot函数来可视化基因区间和元数据。之后,我们找到了两个GRanges对象之间的重叠区间,并对这些区间进行统计摘要。最后,代码通过创建一个查询区间来找出与gr对象重叠的区间,并将查询结果转换为数据框进行分析及可视化。由此我们可以更加坚信无论是进行基因注释、比对分析,还是进行更深入的基因组学研究,GenomicRangesR包都能成为我们的得力助手!
希望本期的内容对你有帮助,期待我们下期再见!最后如果各位小伙伴们觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图呢,附云生信链接
(http://www.biocloudservice.com/home.html)。
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果

往期回顾
|
01 |
|
02 |
|
03 |
|
04 |