想要一键搞定所有单细胞数据的批次效应?除非你用Harmony
 最近,小果发现许多小伙伴在处理多个组学数据集的时候,常常被批次效应困扰,让人感觉头大无比。别担心,小果今天就来为你解困!我要介绍的是一个神奇的R包,它的名字叫做Harmony。这个R包专门用于处理和整合多个组学数据集,极大地减小了批次效应的影响。使用它,你可以自如地整合不同来源、不同批次的数据,让你的研究更加深入和精细。下面就让我们开始吧!
最近,小果发现许多小伙伴在处理多个组学数据集的时候,常常被批次效应困扰,让人感觉头大无比。别担心,小果今天就来为你解困!我要介绍的是一个神奇的R包,它的名字叫做Harmony。这个R包专门用于处理和整合多个组学数据集,极大地减小了批次效应的影响。使用它,你可以自如地整合不同来源、不同批次的数据,让你的研究更加深入和精细。下面就让我们开始吧!
在开始之前,小果先科普一下批次效应(也称技术混杂因素),是指在高通量生物数据分析中,由于不同的实验条件、操作者、分离方法、试剂、仪器、进行实验的实验室、时间等因素导致的与生物变异无关的技术差异。批次效应会影响数据的质量和可信度,干扰数据之间的真实差异,导致分析结果的偏差和误差。因此,消除或减少批次效应是数据分析的一个重要步骤。
Harmony工具包去批次效应的原理:
Harmony 是一种用于单细胞 RNA-测序数据 (scRNA-seq) 的批次效应修正方法。由于它是在单细胞转录组学中使用的,小果简要概括一下它的工作原理。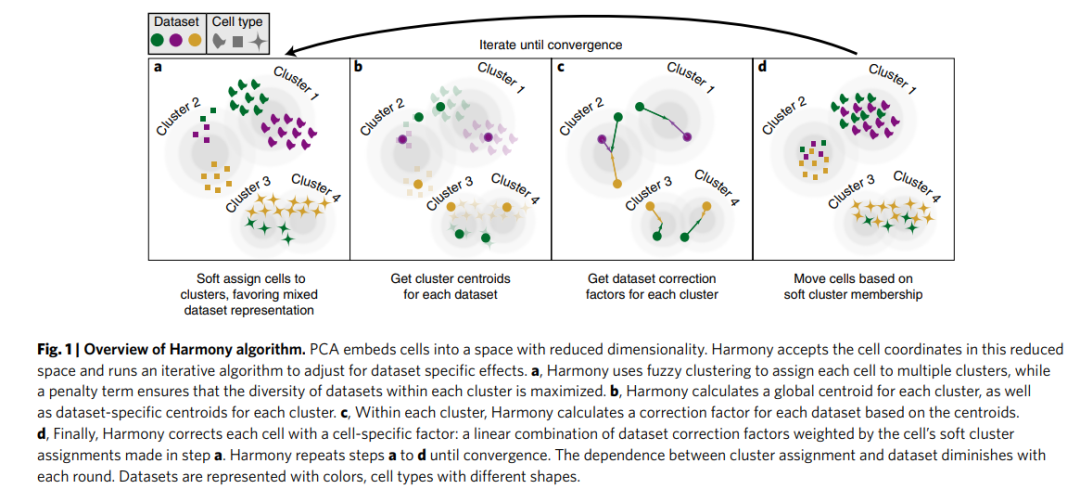
• 首先,使用主成分分析(PCA)将细胞嵌入到低维空间中。
• 然后,使用模糊聚类将每个细胞分配到多个聚类中,同时使用一个惩罚项来最大化每个聚类内数据集的多样性。
• 接着,计算每个聚类的全局质心,以及每个聚类的数据集特定质心。
• 然后,根据质心计算每个数据集的校正因子。
• 最后,使用特定于细胞的因子校正每个细胞:由在模糊聚类中进行的软分配加权的数据集校正因子的线性组合。
重复上述步骤直到收敛。
所以,Harmony 的工作原理就是通过找出不同细胞批次之间的相似基因表达模式,并根据这些模式调整它们的数据,从而消除批次效应。想深入了解的小伙伴可以去看原文:https://www.nature.com/articles/s41592-019-0619-0

首先,让我们来了解一下Harmony包的安装步骤。在R环境中,您可以使用以下代码安装该包:

如果您尚未安装BiocManager,请先安装if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")# 安装Harmony包BiocManager::install("Harmony")
小Tip
假如还安装不上,也可以用小果之前介绍的devtools一键安装
library(devtools)install_github("immunogenomics/harmony")
安装完成后,我们可以开始使用Harmony包来调整批次效应。
实战演练:
下载示例数据(小果已经帮你找好了
百度云链接:
https://pan.baidu.com/s/1BcimNtpmfYPCXAzJDaBaUQ
提取码:lnfy
# 导入数据, 是两个稀疏矩阵
load('pbmc_test.RData')# 导入所需的库
library(harmony)library(Seurat)
#创建一个Seurat对象,用于存储和分析单细胞转录组数据。counts参数是指定计数矩阵,这里是将两个稀疏矩阵(分别代表刺激组和对照组的单细胞数据)按列合并。min.cells=5,表示只保留在至少5个细胞中有表达的基因。
pbmc <- CreateSeuratObject(counts = cbind(stim.sparse, ctrl.sparse), project = "PBMC", min.cells = 5)#给Seurat对象添加新列,将每个细胞分为刺激组或是对照组。
pbmc@meta.data$group <- c(rep("STIM", ncol(stim.sparse)), rep("CTRL", ncol(ctrl.sparse)))#对矩阵进行归一化处理,使每个细胞的基因表达量可以在不同细胞之间进行比较。verbose参数指是否打印出详细信息。
pbmc <- Seurat::NormalizeData(pbmc,verbose = FALSE)#找出不同细胞之间有显著差异的基因。,用于后续的聚类和降维分析。selection.method参数是指定选择方法,这里是”vst”,意思是使用方差稳定化变换(variance stabilizing transformation)。nfeatures参数是指定要选择的特征基因数目,这里是2000。
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)#将所有基因的名称赋值给一个变量
all.genes <- rownames(pbmc)#对变异特征基因进行标准化处理,使每个基因的表达量有一个均值为0,标准差为1的正态分布。这样可以消除不同基因之间的量纲差异,也可以减少异常值的影响。features参数是指定要进行标准化的基因列表,这里是之前存储的所有基因。
pbmc <- ScaleData(pbmc, features = all.genes)#降维聚类
#使用主成分分析(principal component analysis)将高维的数据投影到低维的空间中,形成一组互相正交的主成分(principal components),每个主成分都代表了数据中的一个方差最大的方向。npcs参数是指定要保留的主成分数目,这里是30。
pbmc <- RunPCA(pbmc, npcs = 50)# 根据主成分计算每个细胞与其他细胞之间的相似度,并构建一个邻接矩阵。dims参数是指定要使用的主成分范围,这里是1到30。
pbmc <- FindNeighbors(pbmc, dims = 1:30)# 根据邻接矩阵对细胞进行聚类,也就是将相似度高的细胞划分为一个群体。resolution参数是指定聚类的精度,也就是决定聚类个数的参数,数值越大聚类个数越多。
pbmc <- FindClusters(pbmc, resolution = 0.3)# 对聚类后的数据进行UMAP(uniform manifold approximation and projection)以便于在二维或三维空间中可视化细胞群体。
pbmc <- RunUMAP(pbmc, dims = 1:30)# 按分组和聚类数目绘制组别umap图
p1 <- DimPlot(pbmc, reduction = "umap",group.by = "group", pt.size=0.5, label = F,repel = TRUE)p2 <- DimPlot(pbmc, reduction = "umap",group.by = "seurat_clusters", pt.size=0.5, label = F,repel = TRUE)
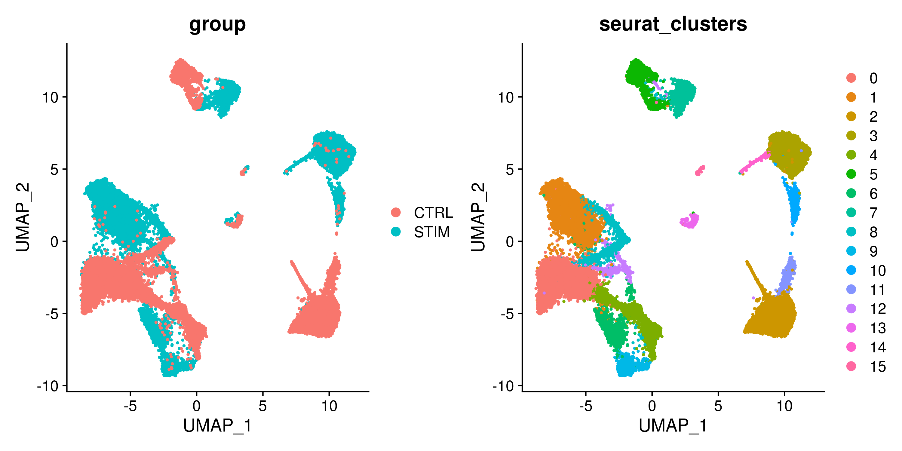
可以发现未经校正的数据集之间存在明显差异
# 使用RunHarmony函数去批次
#按照样本组别去批次,这里使用了管道函数%>%,即把函数左边的值发给右边做后续处理。
pbmc = pbmc %>% RunHarmony("group", plot_convergence = TRUE)

Harmony算法在进行了6次迭代后收敛,此时它找到了一个最优的解决方案,使得数据集之间的差异最小化,而细胞类型之间的差异最大化。
pbmc2 <- pbmc2 %>%RunUMAP(reduction = "harmony", dims = 1:30) %>%FindNeighbors(reduction = "harmony", dims = 1:30) %>%FindClusters(resolution = 0.3) %>%identity()p3 <- DimPlot(pbmc, reduction = "umap",group.by = "group", pt.size=0.5, label = F,repel = TRUE)p4 <- DimPlot(pbmc, reduction = "umap",group.by = "seurat_clusters", pt.size=0.5, label = F,repel = TRUE)
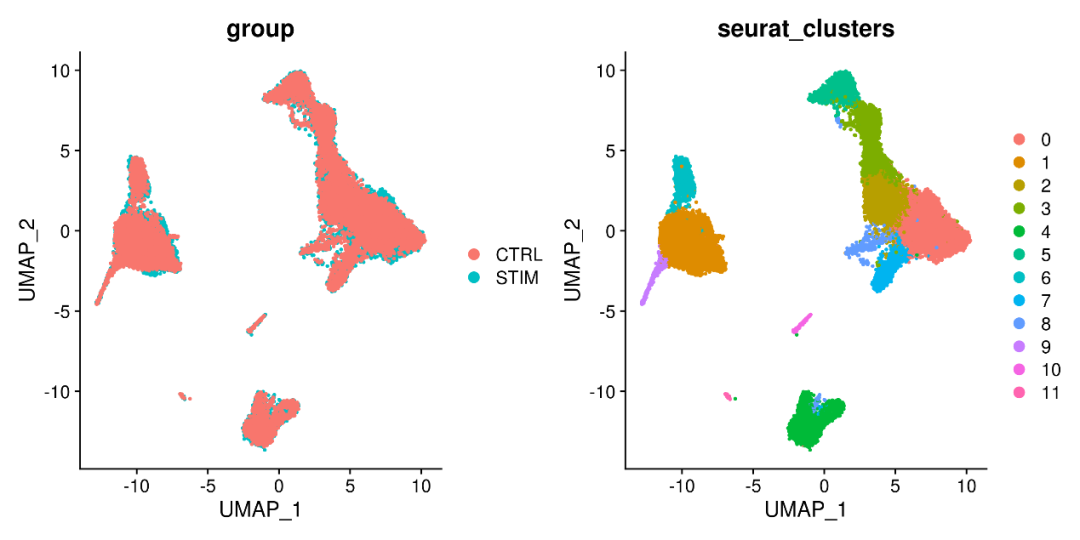
#去批次后样本分布的比较一致
看,是不是非常简单?Harmony包就是这么神奇,可以让你轻松处理和整合多个组学数据集,让你的研究更加顺利。但是,请记住,这只是一个基础的使用教程,Harmony包的强大功能还有很多等着你去发掘。所以,小伙伴们,如果你们手头有多个组学数据集需要处理,那就赶快动手试试吧!
往期推荐

