跟着小果学复现-手把手带你拿下IF=46.9Nature 级别的主成分分析(PCA)图!!

小果最近阅读了许多的文献,心里一直在思考,科研人员如何发表成功的高分的生信文章。最终呢小果虽然没有完全发掘其中的奥秘,但是小果发现一个规律,这些高分文章中对数据的把控是很到位,如何理解呢,小果看的Nature级别的文章中,他们都数据量都很庞大,
所以小果就想说,小伙伴如果想发高分文章一开始处理的数据一定不能少,样本量够大,这样也可以侧面的说明工作量大,那么要是解决这个问题,后续的思路等就迎刃而解了。
说到数据,小伙伴做分析要是用很大的数据,那就少不了对数据的质控。那该如何指控呢,小果告诉小伙伴用的最多的就是主成分分析(PCA),
目前许多的生信分组虽然他们经常会做PCA分析,但是他们都是很普通的可视化方式,最多的就是横纵坐标的PCA二维图,再者就是三维的PCA图。小果想教给小伙伴不一样方式的PCA图,然后就发现一篇Nature文章中的PCA图,这幅图打破正常我们见过的PCA图,我们先看图在了解一下这个图的特色,这是来自IF影响因子有46.9《Natue biotechnology》中的《Removing unwanted variation from large-scale RNA sequencing data with PRPS》图2a,来看一下:
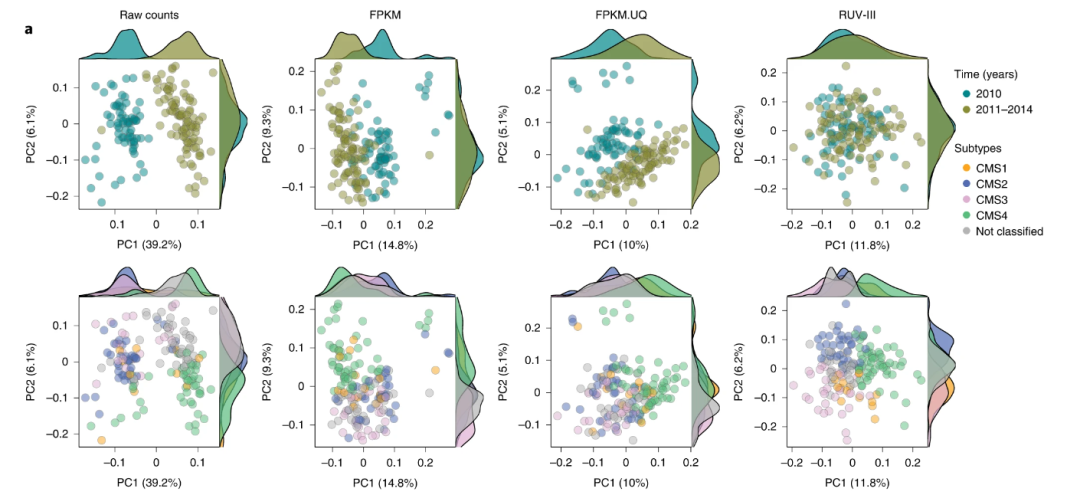
小伙伴看的第一眼是不是觉得这不是PCA图,小果也是第一眼就被吸引了,这幅图,PCA主要的内容应有尽有,并且还非常的好看,那么今天小果就带小伙伴去复现这张图,我们以后做PCA就可以用到这种图了,
作图的前提呢,小果还是带小伙伴了解一下什么是PCA,我们也要明白为什么要做这个图,其实主成分分析属于一种降维算法,当我们的数据量很庞大的时候就需要去把数据降维,而降维呢就是一种对高维度特征数据预处理方法。降维是将高维度的数据保留下最重要的一些特征,去除噪声和不重要的特征,从而实现提升数据处理速度的目的。
在实际的生产和应用中,降维在一定的信息损失范围内,可以为我们节省大量的时间和成本。降维也成为应用非常广泛的数据预处理方法。
这么做是有利于我们分析的,可以使得数据集更易使用。也可以使得结果容易理解。
那么话不多说,我们就开始下面的学习吧!
这里呢小果还要说明一下,我们做PCA,数据库很大的话,那么我们操作占用内存比较大,所以建议各位小伙伴可以使用服务器去运行,如果没有自己的服务器也没有关系,可以欢迎联系我们使用服务器租赁~
 好了,我们开始吧!
好了,我们开始吧!
我们在做PCA分析的时候,因为对许多数据进行处理,这就需要用到一些函数去计算,
这次小果就分享给小伙伴两个函数,来实现我们这次的复现内容,
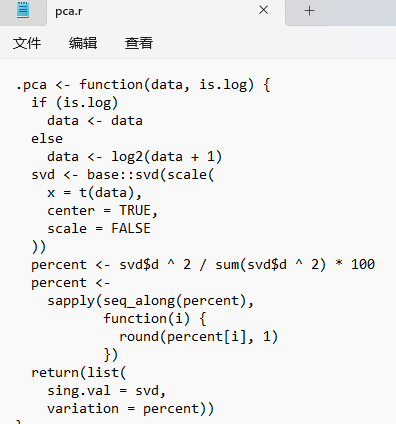
第一个就是我们用来计算主成分分析的pca函数
.pca <- function(data, is.log) {if (is.log)data <- dataelsedata <- log2(data + 1)svd <- base::svd(scale(x = t(data),center = TRUE,scale = FALSE))percent <- svd$d ^ 2 / sum(svd$d ^ 2) * 100percent <-sapply(seq_along(percent),function(i) {round(percent[i], 1)})return(list(sing.val = svd,variation = percent))}这个函数可以计算我们的所有的数据,小伙伴也不用理解这个函数具体意思,
第二个函数就是用来作图的,这个就需要用到我们的老朋友ggplot2包中的ggpubr还有cowplot包,
来看一下:
.scatter.density.pc <- function(pcs,pc.var,group.name,group,color,strokeSize,pointSize,strokeColor,alpha,title){pair.pcs <- utils::combn(ncol(pcs), 2)pList <- list()for(i in 1:ncol(pair.pcs)){if(i == 1){x <- pair.pcs[1,i]y <- pair.pcs[2,i]p <- ggplot(mapping = aes(x = pcs[,x],y = pcs[,y],fill = group)) +xlab(paste0('PC', x, ' (', pc.var[x], '%)')) +ylab(paste0('PC', y, ' (', pc.var[y], '%)')) +geom_point(aes(fill = group),pch = 21,color = strokeColor,stroke = strokeSize,size = pointSize,alpha = alpha) +scale_x_continuous(breaks = scales::pretty_breaks(n = 5)) +scale_y_continuous(breaks = scales::pretty_breaks(n = 5)) +ggtitle(title) +theme(legend.position = "right",panel.background = element_blank(),axis.line = element_line(colour = "black", size = 1.1),legend.background = element_blank(),legend.text = element_text(size = 12),legend.title = element_text(size = 14),legend.key = element_blank(),axis.text.x = element_text(size = 10),axis.text.y = element_text(size = 10),axis.title.x = element_text(size = 14),axis.title.y = element_text(size = 14)) +guides(fill = guide_legend(override.aes = list(size = 4))) +scale_fill_manual(name = group.name, values = color)le <- ggpubr::get_legend(p)}else{x <- pair.pcs[1,i]y <- pair.pcs[2,i]p <- ggplot(mapping = aes(x = pcs[,x],y = pcs[,y],fill = group)) +xlab(paste0('PC', x, ' (',pc.var[x], '%)')) +ylab(paste0('PC', y, ' (',pc.var[y], '%)')) +geom_point(aes(fill = group),pch = 21,color = strokeColor,stroke = strokeSize,size = pointSize,alpha = alpha) +scale_x_continuous(breaks = scales::pretty_breaks(n = 5)) +scale_y_continuous(breaks = scales::pretty_breaks(n = 5)) +theme(panel.background = element_blank(),axis.line = element_line(colour = "black", size = 1.1),legend.position = "none",axis.text.x = element_text(size = 10),axis.text.y = element_text(size = 10),axis.title.x = element_text(size = 14),axis.title.y = element_text(size = 14)) +scale_fill_manual(values = color, name = group.name)}p <- p + theme(legend.position = "none")xdens <- cowplot::axis_canvas(p, axis = "x")+geom_density(mapping = aes(x = pcs[,x],fill = group),alpha = 0.7,size = 0.2) +theme(legend.position = "none") +scale_fill_manual(values = color)ydens <- cowplot::axis_canvas(p,axis = "y",coord_flip = TRUE) +geom_density(mapping = aes(x = pcs[,y],fill = group),alpha = 0.7,size = 0.2) +theme(legend.position = "none") +scale_fill_manual(name = group.name, values = color) +coord_flip()p1 <- insert_xaxis_grob(p,xdens,grid::unit(.2, "null"),position = "top")p2 <- insert_yaxis_grob(p1,ydens,grid::unit(.2, "null"),position = "right")pList[[i]] <- ggdraw(p2)}pList[[i+1]] <- lereturn(pList)}
小伙伴可以直接运行这两个函数,或者也可以想小果一样整理在一个R文件里面,方便下次使用,
然后我们就可以使用
source(“pca.r”)#载入使用
下面我们就是来看下载入数据的格式,
这是小图自己的数据,小伙伴可以按照形式自行去设置,
df<-read.csv("pca1.csv" )
df
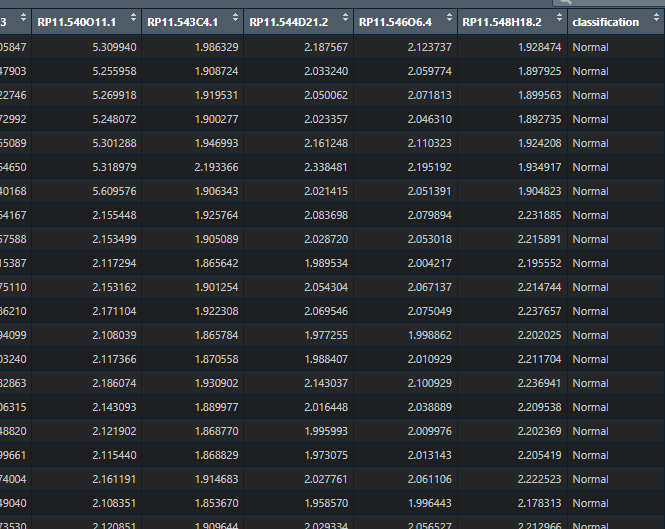
我们展示最重要的一部分,前面都是基因的表达量,纵的表示样本,最后一个我们设置一下分组,作为我们PCA分析的数据形式,就是Classification,小图这里分了四组,文献中分了5组,这个看小伙伴自己的需求,
然后我们计算一下
pca.ncg<-.pca(data = df[,1:8767], ##is.log = FALSE)
上面这个数字代表基因数量,小图这里就选择这一些
运行完就
 出现,我们就将计算好的值进行可视化,
出现,我们就将计算好的值进行可视化,
.scatter.density.pc(pcs = pca.ncg$sing.val$v[, 1:3], ##展示几个pc.var = pca.ncg$variation,strokeColor = 'gray30',strokeSize = .2,pointSize = 2,alpha = .6,title = "A", ##标题名group.name = "Stage", # legned namegroup=df$classification, # 选择分组color=c("darkorchid4","cornflowerblue","sandybrown","mediumaquamarine")) -> p#颜色下面我们使用代码进行展示,do.call(gridExtra::grid.arrange,c(p,ncol=4,nrow=1))
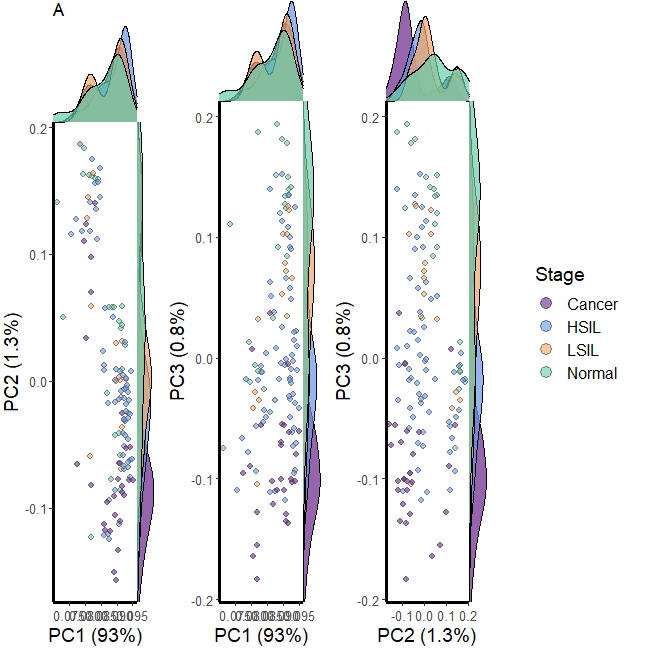
这样一看,和文献中有点不一样,太长了,我们把出图的排列改一下
do.call(gridExtra::grid.arrange,c(p,ncol=2,nrow=2))
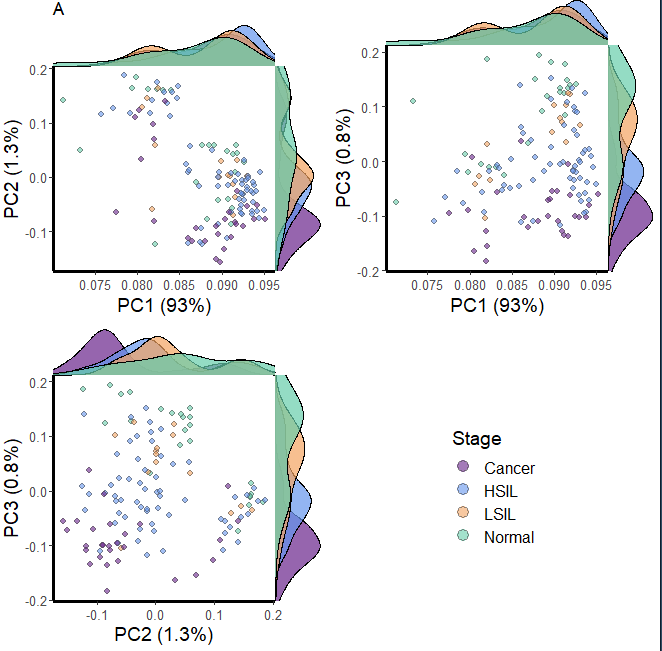
这样一来就差不多了,最后再把点的大小调一下,然后出图,
.scatter.density.pc(pcs = pca.ncg$sing.val$v[, 1:3], ##pc.var = pca.ncg$variation,strokeColor = 'gray30',strokeSize = .2,pointSize = 4,alpha = .6,title = "A", ##group.name = "Stage", # legned namegroup=df$classification, #color=c("darkorchid4","cornflowerblue","sandybrown","mediumaquamarine")) -> pdo.call(gridExtra::grid.arrange,c(p,ncol=2,nrow=2))
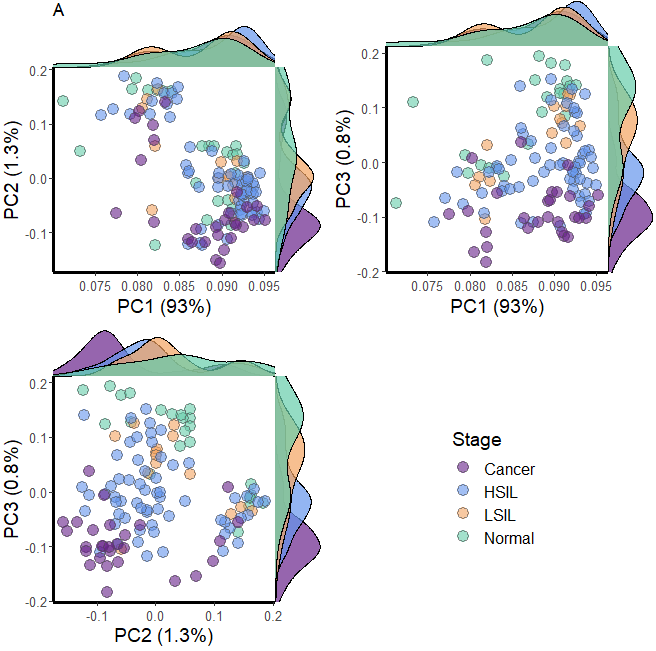
这就一样了基本上就一样了,颜色小图就随机设置的,小伙伴看了有没有心动了,要多多理解代码的意思,上述函数的文件呢,小伙伴需要的可以找我们公众号领取哦
最后呢,小果给小伙伴带个小福利,如果小伙伴觉得自己运行代码太麻烦,可以试试我们的云生信小工具哟,里面有成熟的代码,只要输入合适的数据就可以直接出想要的图呢。这是我们的网址http://www.biocloudservice.com/home.html
多多关注我们公众号,一来学生信!


往期推荐