一学就会的拟时序分析:用Monocle2洞察细胞发育

亲爱的果粉儿们,你们好,小果又来啦!小果今天给大家带来的分享是如何使用Monocle2进行单细胞RNA-seq数据分析。这个分析工具在单细胞研究中非常实用,小果强烈推荐小伙伴们学习。一起跟随小果的步伐开始今天的学习吧!

Monocle2是何方神圣?
Monocle2是一个强大的R包,它可以帮助我们分析单细胞RNA-Seq数据,研究细胞的发育轨迹、分化状态、转录调控特征等等。用它,你就可以像看电影一样,看到细胞的生命历程。迄今为止,Monocle已经发布到了第三代,而Monocle2是目前做单细胞拟时序分析最有名的R包。相较于第一代它有有更丰富的功能,而相较仍在持续开发中的Monocle3来说,Monocle2更稳定且更倾向于半监督的分析模式,可以利用先验知识或标记基因来进行个性化的轨迹分析。
什么是拟时序分析?
拟时序分析是一种利用单细胞RNA-Seq技术来测量每个细胞的基因表达水平,从而揭示细胞的异质性、功能和发育过程的方法。拟时序分析可以帮助我们回答很多有趣的生物学问题,比如:
• 细胞是如何从一个原始状态发育成不同的类型和功能的?
• 细胞之间有哪些差异和相似性?
• 细胞在不同的条件下会如何表达不同的基因?
• 细胞在不同的时间点会如何变化和演化?
为了回答这些问题,我们需要用到monocle2这个强大的R包。monocle2可以帮助我们做以下几件事情:
• 对单细胞数据进行质量控制、标准化、降维和聚类。
• 对单细胞数据进行轨迹分析,即根据基因表达水平来排序和连接每个细胞,构建出一个代表细胞发育过程的图。
• 对单细胞数据进行差异表达分析,即找出在不同轨迹阶段或不同细胞类型之间表达差异显著的基因。
• 对单细胞数据进行可视化,即用各种图形来展示轨迹、聚类、差异表达等结果。
听起来很酷吧?那么让我们开始吧!首先,我们需要安装monocle2这个R包。
如何使用Monocle2?
首先,我们需要在R环境中安装Monocle2包
BiocManager::install("monocle")library(monocle)
然后,我们就可以使用Monocle2来分析单细胞数据了。
加载其他需要的库
library(dplyr)library(Seurat)library(patchwork)
准备数据
测试数据来自10X Genomics免费提供的外周血单核细胞(PBMC)数据集。将近2700个细胞。小果已经提前进行标准化处理,并注释好了细胞类型,储存为rds文件。获取方式如下。
链接:https://pan.baidu.com/s/1SsrPcXyXX9qb9MtiLlIDRQ?pwd=x3mm
提取码:x3mm
#导入注释好的Seurat对象pbmc <- readRDS("pbmc3k_test.rds")#进行逆时序分析需要准备三个文件:表达矩阵文件(exprs)、基因属性文件(featureData)和细#胞属性文件(phenoData),这三个文件都可以从我们处理好的Seurat对象中获取。#表达矩阵,以稀疏格式处理数据,可以加快计算速度exprs <- as(as.matrix(pbmc@assays$RNA@counts), 'sparseMatrix')#基因属性文件,这是一个数据框,每一行代表一个基因,每一列代表一个属性,比如基因#名、基因长度等。featureData <- data.frame(gene_short_name = row.names(pbmc),row.names = row.names(pbmc))fd <- new('AnnotatedDataFrame', data = featureData)#细胞属性文件,这是一个数据框,每一行代表一个细胞,每一列代表一个属性,比如细胞#类型、样本来源等。phenoData <- pbmc@meta.dataphenoData$celltype <- pbmc@active.identpd <- new('AnnotatedDataFrame', data = phenoData)
创建对象
#准备好了我们就可以开始用monocle2的newCellDataSet函数来创建一个单细胞数据对象,#这个对象会存储所有的数据和分析结果,用于后续monocle分析
cds <- newCellDataSet(exprs,phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())#估计尺寸因子和分散度,这一步非常关键,不能跳过#分别有助于我们对跨细胞捕获的mRNA差异进行归一化和后续差异表达分析。cds <- estimateSizeFactors(cds)cds <- estimateDispersions(cds)##过滤低质量细胞,过滤低于1%细胞中检出的基因cds <- detectGenes(cds, min_expr = 0.1)#筛选出在至少10个细胞中有表达的基因#从对象中排除在少数细胞中表达的基因,以免浪费CPU时间分析它们的差异表达。expressed_genes <- row.names(subset(fData(cds),num_cells_expressed >= 10))
细胞分类
Monocle 提供了一个简单的系统,用于根据我们选择的标记基因的表达来鉴定#细胞。但是术业有专攻,monocle毕竟不是专门作细胞鉴定的软件,小果还是建议先做好细#胞注释以后再导入monocle进行分析,贴心的小果今天就提前处理了,吼吼~
构建单细胞轨迹
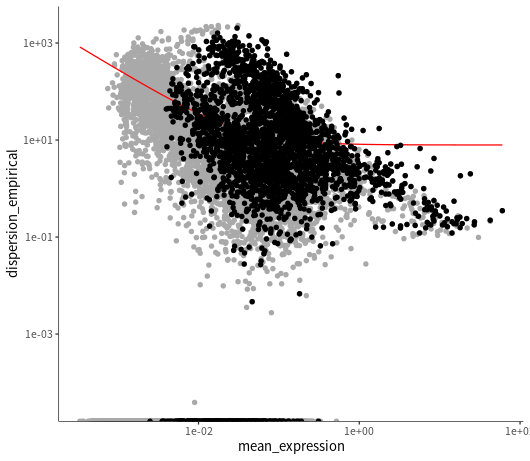
# 第1步:选择定义细胞进展的基因#对cds对象中的expressed_genes基因进行差异表达分析,并将结果赋值给diff变量。~ celltype表示我们想要比较不同细胞类型之间的基因表达差异diff <- differentialGeneTest(cds[expressed_genes,],fullModelFormulaStr="~celltype",cores=1)#找出那些在不同细胞类型之间有显著差异表达的基因,以用于细胞排序。ordergene <- rownames(subset(diff, qval < 0.01))#标记为用于细胞排序的基因#一旦我们有了用于排序的基因 ID 列表,我们需要在 cds对象中设置它们。cds <- setOrderingFilter(cds, ordergene)#其中黑点就是后面构建发育轨迹需要的基因plot_ordering_genes(cds)
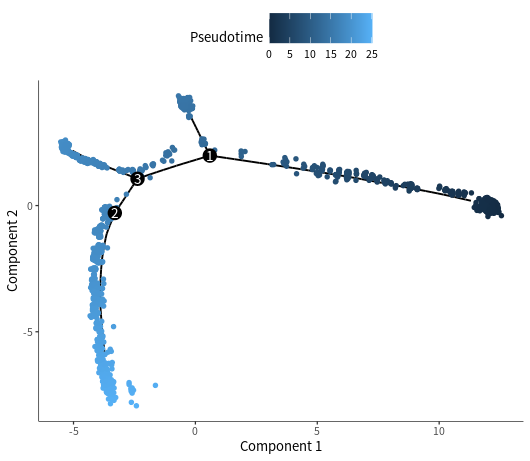
#第二步: 降低数据维度接下来,我们降维到二维空间,我们将能够在Monocle细胞排序时进行可视化和解释。cds <- reduceDimension(cds, max_components = 2,method = 'DDRTree')# 第三步: 沿轨迹对细胞进行排序cds <- orderCells(cds)#按Pseudotime展示,Pseudotime是一种用来衡量每个细胞在发育轨迹中的相对位置或虚拟时间的指标,它反映了每个细胞的发育进程和分化状态。Pseudotime可以帮助我们发现那些随着发育过程而变化的基因或转录因子。plot_cell_trajectory(cds,color_by="Pseudotime", size=1,show_backbone=TRUE)
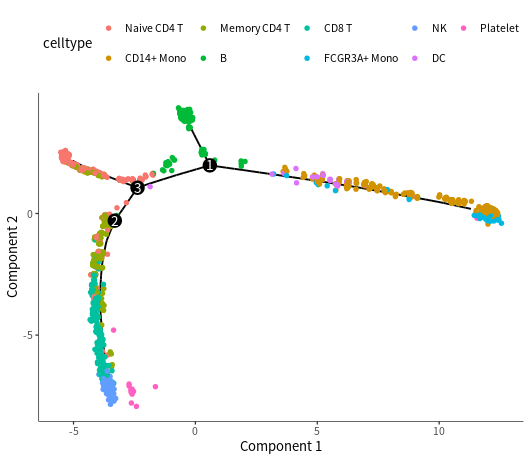
#按细胞类型展示plot_cell_trajectory(cds,color_by="celltype", size=1,show_backbone=TRUE)
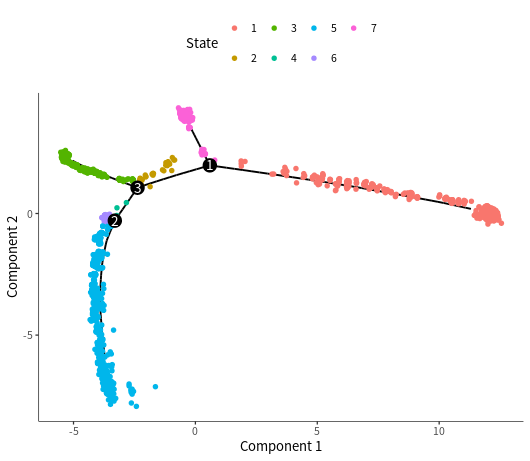
#按state展示,还可以作分面图plot_cell_trajectory(cds, color_by = "State",size=1,show_backbone=TRUE)plot_cell_trajectory(cds, color_by = "State") + facet_wrap("~State", nrow = 1)
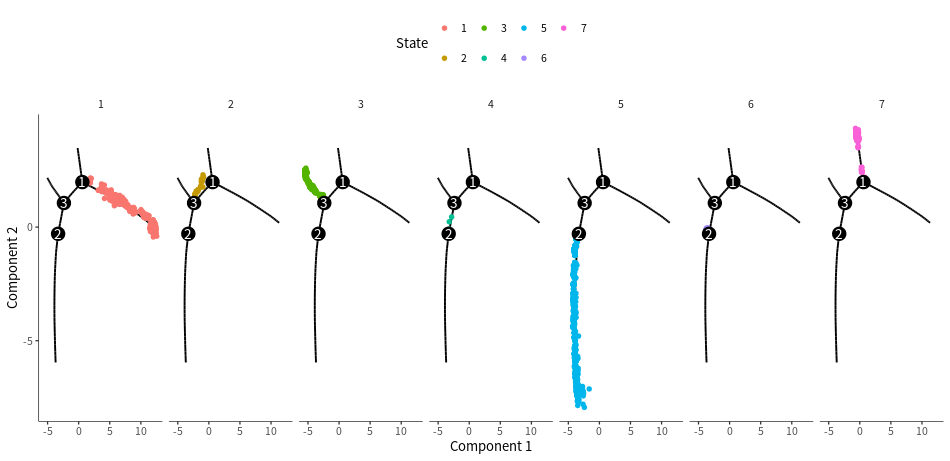
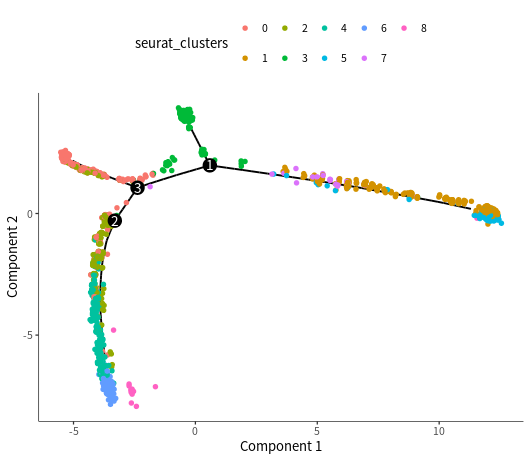
#按注释之前的分群结果展示plot_cell_trajectory(cds, color_by = "seurat_clusters")
今天,小果就带大家探索到这里!希望你已经简单了解如何使用Monocle2来观察单细胞的发展历程。如果有什么问题或者疑问,欢迎在评论区留言,小果会秒回的哦。如果你觉得今天的内容有用,别忘了点赞和分享哦,这是对小果的最大支持!我们下次再见!
