DNA生信分析(一):萌新必看!一分钟学会如何处理FASTQ文件
点击蓝字 关注我们
一分钟学会如何处理FASTQ文件
在前面的文章中,小花向大家分享了单细胞测序的流程,想必小伙伴们也成功的学会了湿实验的步骤吧。但是湿实验的结束才是整个生信项目的开始,面对着测序仪下机的原始数据,很多小伙伴可能感觉无从下手,不知道怎么对数据进行分析。这一次小花就跟大家分享一下怎么阅读从测序仪得到的FASTQ文件,大家快来看看吧!
什么是FASTQ文件?
FASTQ(Fast Quality)文件是一种常用的存储测序数据的文本格式,文件后缀通常为.fastq或.fq。它通常用于存储高通量测序(如DNA测序或RNA测序)得到的原始测序片段(reads)的序列和对应的质量信息。
FASTQ文件的每个测序片段通常由四行组成,具体格式如下:
1.第一行以符号 “@” 开头,后接一个唯一的标识符,表示该序列的名称或编号。
2.第二行是对应测序片段的碱基序列,由A、T、C、G等碱基字符组成。
3.第三行以符号 “+” 开头,通常跟随着与第一行相同的标识符,用于可选的描述性信息(在一些情况下可以忽略)旧版FASTQ格式中会在这一行重复第一行信息,现在为节约存储,一般是不再加重复信息。
4.第四行是与第二行对应的质量值序列,这一行信息至关重要,通常使用ASCII编码表示,每个字符对应一个测序片段中对应位置的质量值。
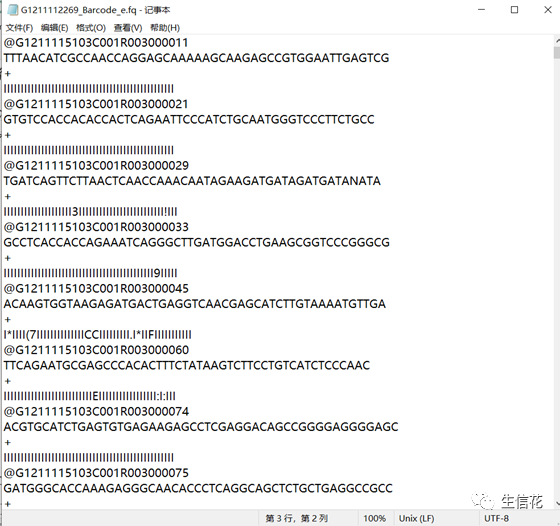
这个FASTQ文件是小果在一次DNA测序后从测序仪直接得到的下机数据。以这个文件为例,小伙伴们是不是已经理解了每个片段的前三行呢?接下来小花就详细给大家解释一下第四行质量值的阅读方法。
什么是质量值Q和质量分数Phred?
质量值Q表示了测序片段中每个碱基的可信度,碱基质量值Q计算公式:

其中p为碱基被识别错误的概率。根据公式计算所得Q值越高,代表碱基被测错的概率越小。例如测序数据下机质量评价指标中,Q20,Q30,Q40就分别代表测序错误率为1%,0.1%,0.01%。下机数据Q30 >85%,代表所有下机数据当中测序错误率小于0.1%的碱基占总碱基数的比例超过85%。
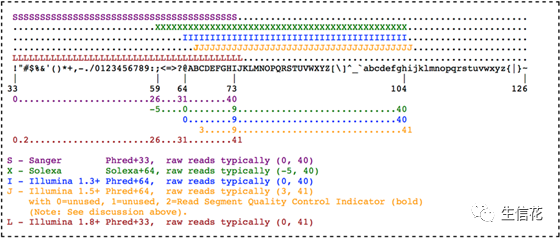
Phred质量分数是一个负数,代表着错误的概率,一般在0到41之间(ASCII编码的范围是33-73),较低的质量分数表示较高的错误概率。
为了对碱基质量值进行准确记录并节约存储,采用ASCII表对碱基质量值进行转换。对应转换方式比较常见的有Phred33和Phred64两种不同的质量体系。
Phred33:从ASCII码表可打印的字符开始,将质量值0~40对应转换为ASCII值33到73对应的控制字符“!”~“I”:(即用“实际碱基质量值+33”所得数值对应的ASCII码表中的控制字符代表该碱基质量值)
Phred64:用“实际碱基质量值+64” 所得数值对应的ASCII码表中的控制字符代表该碱基质量值。
其他质量值体系如Solexa+64等的质量值与控制字符的对应关系见以下图示。
需要注意的是,由于FASTQ文件的体积通常较大,在处理和传输时需要考虑存储空间和计算资源的限制。
看到这里,小花相信大家已经掌握了如何阅读FASTQ文件,但是可能还有小伙伴们会有疑问,FASTQ文件可以进行什么处理呢?能够变成什么文件呢?与其他文件类型有什么关系呢?不要心急,小果这就给大家娓娓道来。
FASTQ文件后续如何处理?
初入生信的小伙伴们可能听说过FASTA文件(.fasta/.fa),与FASTQ名称有八成甚至九成的相似,可能很多小伙伴都不太能分清他们,现在就让小果用“照妖镜”揭开FASTQ文件的朋友们的神秘面纱吧:
举一个FASTA文件的例子:

相信聪明的小伙伴们已经看出来了,其实FASTA文件就是FASTQ文件的序列值部分,即FASTQ文件每个序列的前两行。正如上面所说,由于FASTQ文件的体积通常较大,在处理和传输时需要考虑存储空间和计算资源的限制,因此FASTQ文件经常需要转化成体积更小的FASTA文件。将FASTQ转换为FASTA格式,即把FASTQ中的序列行取出的过程就叫做序列拼接。这样得到了忽略质量值的FASTA文件,是为了适用于某些序列分析和比对算法。
那么FASTA文件又可以如何接着处理呢?在本系列教程的后面,我们还会继续接触序列比对、变异检测等等操作,这些操作也都会生成各种文件类型,小花在这里就一次性满足小伙伴们的好奇心,继续往下展开分享吧~
使用短序列比对算法(例如Bowtie、BWA等)将FASTQ文件比对到参考基因组或其他序列库上,生成的比对结果文件通常是SAM(Sequence Alignment/Map)或BAM(Binary Alignment/Map)格式。这些文件包含了每个测序 read 的比对位置、序列以及其他相关信息。SAM文件是文本格式,BAM文件是其二进制形式。SAM/BAM文件的主要应用是存储和传输测序数据比对到参考基因组(或转录组)的结果。这些文件包含了每个测序read的比对位置、比对质量、序列信息等重要信息。关于SAM/BAM文件的详细信息,小花会在后面的教程中给大家娓娓道来。
到这里还远远不是生信分析的尽头。SAM/BAM文件是存储测序数据比对结果的标准格式,广泛应用于测序数据分析、基因组注释和变异检测等领域。例如:
1.对比对结果进行变异检测时,可以将SAM/BAM文件与参考基因组进行比较,识别出测序样本中与参考基因组不同的变异位点,并生成Variant Call Format(VCF)文件。
2.根据SAM/BAM文件的覆盖程度和特征,可以生成BED(Browser Extensible Data)文件,用于表示基因组区域的注释、染色体结构变异等信息。
3.将比对结果(SAM/BAM文件)与注释数据库(如基因组注释数据库)结合,生成GTF(Gene Transfer Format)或GFF(General Feature Format)文件,用于描述基因和转录本的结构、外显子、内含子等信息。
4.为了快速访问和检索BAM文件中的比对结果,可以对BAM文件进行索引构建。索引文件提供了一种快速查询特定区域的机制,避免了需要逐行扫描整个BAM文件的时间消耗。目前常见的BAM索引文件格式有.BAI(Binary Alignment/Map Index)和.CSI(Coordinate-sorted Binary Index)两种格式。
为了方便大家记忆,小花自制一张思维导图,欢迎大家收藏哦~
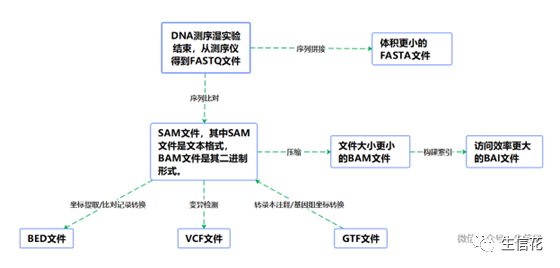
顺带一提,小花在可视化时喜欢使用IGV软件,一般就需要用到.BAI文件。后面小花也会专门出一期教程给大家分享如何使用IGV,小伙伴们记得持续关注哦。
怎么样,相信聪明的大家已经掌握了怎么处理FASTQ文件的内容,是不是很简单呢?其实生信中很多东西都是这样,第一次接触时看上去有点复杂,但只要稍加学习和思考就可以轻易掌握,小花相信大家只要在生信这条路上坚持地走下去,不远的未来都可以成为生信大牛!相信小伙伴们已经摩拳擦掌,迫不及待地想要开启生信分析流程啦,大家可以继续关注小花接下来的内容,小果会全面并且详细地分享生信分析接下来的流程哦。
如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小花开发的生信工具平台http://www.biocloudservice.com/home.html,大家在新接触一个知识的时候,与其先花费大量时间死啃知识点,不如先利用好工具先自己上手接触流程,在跑完一遍全流程后再返回去理解知识点,相信可以更好更快地理解,达到事半功倍的效果!
下一次,小花就跟大家分享如何在五分钟内完成FASTQ文件的质量评估,小伙伴们记得持续关注哦。


(点击阅读原文跳转)
![]() 点一下阅读原文了解更多资讯
点一下阅读原文了解更多资讯
