前言
空间转录组学分析,是近几年的一个热点。相信小伙伴们对于seurat再熟悉不过了!但是seurat是基于R语言生态来进行数据处理的,数据处理起来实在是太麻烦了!Python在图像识别、空间分割和去卷积等算法领域拥有绝对的优势!Scanpy是基于python的单细胞分析工具包,江湖地位相当于R语言中的seurat。重要程度不用我多说了吧!没接触过python的小伙伴们也不要害怕哦!它与seurat的分析流程基本一致,都是质控-选择高变基因-PCA降维-聚类-UMAP(tSNE)降维这些步骤。没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找小云哦!练了十年生信分析的小云对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!
生信数据处理起来占用内存实在太大了,放过自己的电脑吧!小云在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系小云,保证服务器的性价比最高哦!
代码教程
1.导入相关的库
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
sc.logging.print_versions()
sc.set_figure_params(facecolor=”white”, figsize=(8, 8))
sc.settings.verbosity = 3
2.读取数据
我们将使用人类淋巴结的 Visium 空间转录组学数据集。函数 datasets.visium_sge() 从 10x Genomics 下载数据集并返回一个包含计数、图像和空间坐标的对象。使用您自己的 Visium 数据时,请使用 sc.read_visium() 函数导入它。
adata = sc.datasets.visium_sge(sample_id=”V1_Human_Lymph_Node”)
adata.var_names_make_unique()
adata.var[“mt”] = adata.var_names.str.startswith(“MT-“)
sc.pp.calculate_qc_metrics(adata, qc_vars=[“mt”], inplace=True)
adata
3. 质量控制和预处理
我们根据总数和表达基因对样本进行一些基本的过滤
fig, axs = plt.subplots(1, 4, figsize=(15, 4))
sns.histplot(adata.obs[“total_counts”], kde=False, ax=axs[0])
sns.histplot(
adata.obs[“total_counts”][adata.obs[“total_counts”] < 10000],
kde=False,
bins=40,
ax=axs[1],
)
sns.histplot(adata.obs[“n_genes_by_counts”], kde=False, bins=60, ax=axs[2])
sns.histplot(
adata.obs[“n_genes_by_counts”][adata.obs[“n_genes_by_counts”] < 4000],
kde=False,
bins=60,
ax=axs[3],
)
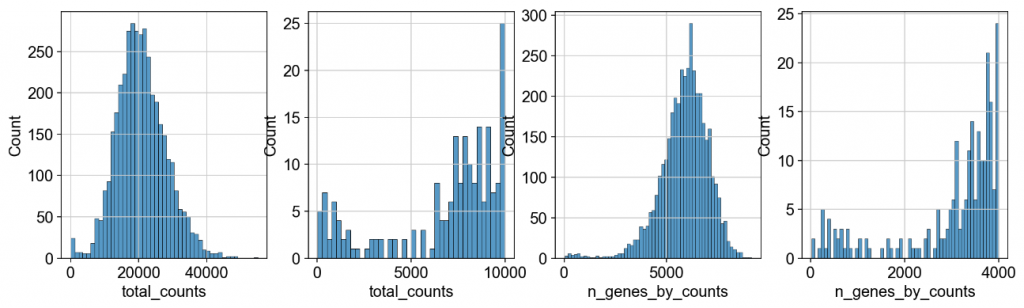
sc.pp.filter_cells(adata, min_counts=5000)
sc.pp.filter_cells(adata, max_counts=35000)
adata = adata[adata.obs[“pct_counts_mt”] < 20].copy()
print(f”#cells after MT filter: {adata.n_obs}”)
sc.pp.filter_genes(adata, min_cells=10)
我们继续使用 Scanpy 的内置方法对 Visium 计数数据进行归一化,并检测高度可变的基因
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor=”seurat”, n_top_genes=2000)
4. 基于转录相似性的流形嵌入和聚类
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(
adata, key_added=”clusters”, flavor=”igraph”, directed=False, n_iterations=2
)
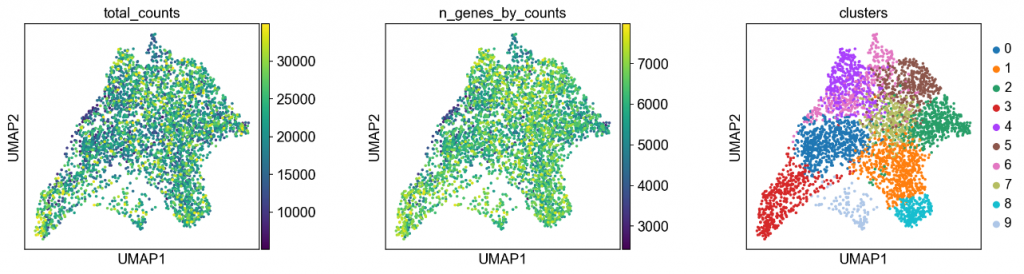
我们绘制了一些协变量,以检查 UMAP 中是否存在与总数和检测到的基因相关的任何特定结构。
plt.rcParams[“figure.figsize”] = (4, 4)
sc.pl.umap(adata, color=[“total_counts”, “n_genes_by_counts”, “clusters”], wspace=0.4)
5. 空间坐标中的可视化
plt.rcParams[“figure.figsize”] = (8, 8)
sc.pl.spatial(adata, img_key=”hires”, color=[“total_counts”, “n_genes_by_counts”])

函数 sc.pl.spatial 接受 4 个附加参数:
- img_key:其中 IMG 存储在元素中adata.uns
- crop_coord:用于裁剪的坐标(左、右、上、下)
- alpha_img:图像透明度的 alpha 值
- bw:标志将图像转换为灰度
之前,我们在基因表达空间中进行了聚类,并用UMAP可视化了结果。通过在空间维度上可视化聚类样本,我们可以深入了解组织组织,并可能深入了解细胞间通讯。
sc.pl.spatial(adata, img_key=”hires”, color=”clusters”, size=1.5)
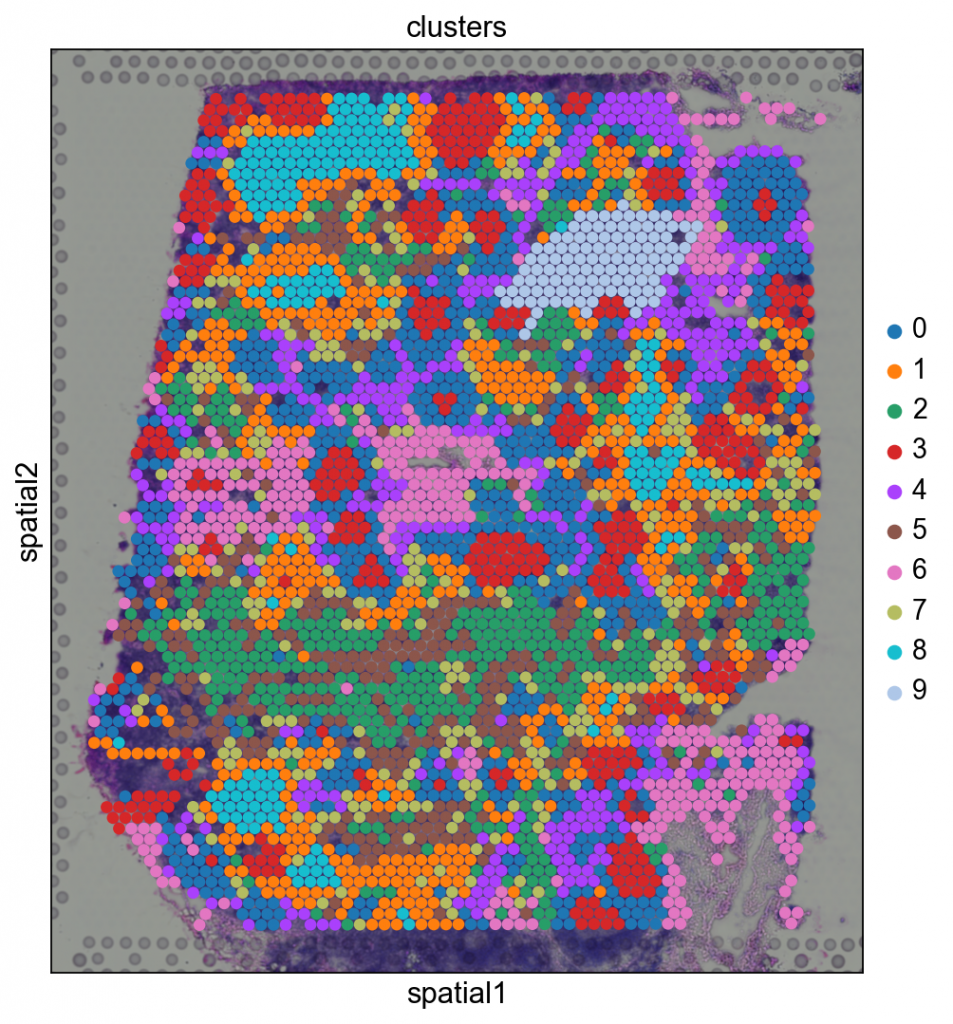
基因表达空间中属于同一簇的斑点通常在空间维度上共现。例如,属于聚类 5 的斑点通常被属于聚类 0 的点包围。
我们可以放大特定的感兴趣区域以获得定性见解。此外,通过改变斑点的 alpha 值,我们可以从 H&E 图像中更好地可视化下面的组织形态。
sc.pl.spatial(
adata,
img_key=”hires”,
color=”clusters”,
groups=[“5”, “9”],
crop_coord=[7000, 10000, 0, 6000],
alpha=0.5,
size=1.3,
)

6. 亚群标记基因
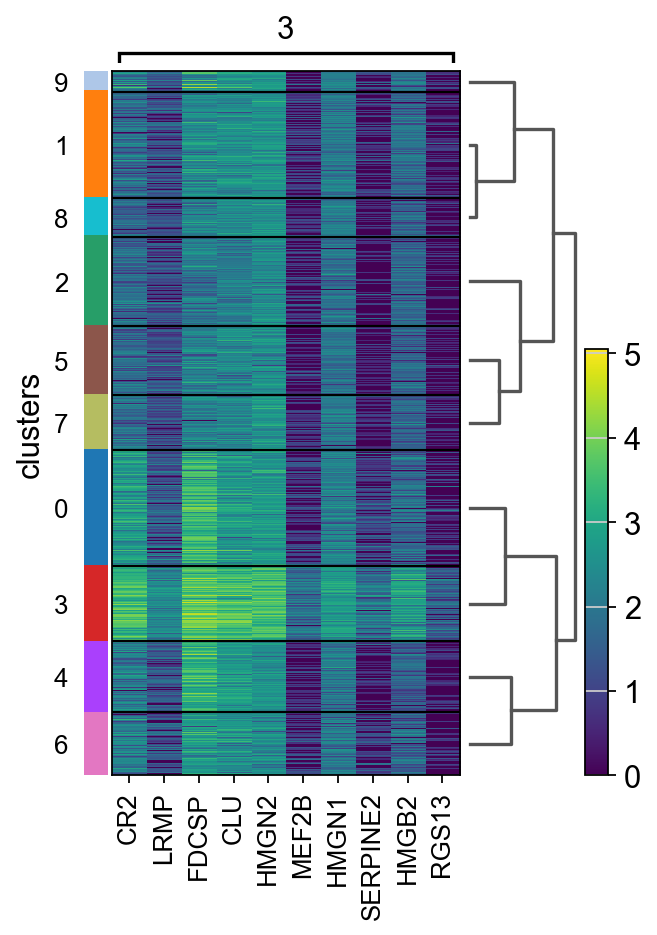
计算标记基因并绘制热图,其中前 10 个标记基因的表达水平。
sc.tl.rank_genes_groups(adata, “clusters”, method=”t-test”)
sc.pl.rank_genes_groups_heatmap(adata, groups=”9″, n_genes=10, groupby=”clusters”)
##我们看到 CR2 概括了空间结构。
sc.pl.spatial(adata, img_key=”hires”, color=[“clusters”, “CR2”])

sc.pl.spatial(adata, img_key=”hires”, color=[“COL1A2”, “SYPL1”], alpha=0.7)

7. MERFISH示例
如果您使用基于 FISH 的技术生成空间数据,只需阅读 cordinate 表并将其分配给元素即可。
# If needed:
# %pip install openpyxl
coordinates = pd.read_excel(“./data/pnas.1912459116.sd15.xlsx”, index_col=0)
counts = sc.read_csv(“./data/pnas.1912459116.sd12.csv”).transpose()
adata_merfish = counts[coordinates.index, :].copy()
adata_merfish.obsm[“spatial”] = coordinates.to_numpy()
#预处理和降维
sc.pp.normalize_per_cell(adata_merfish, counts_per_cell_after=1e6)
sc.pp.log1p(adata_merfish)
sc.pp.pca(adata_merfish, n_comps=15)
sc.pp.neighbors(adata_merfish)
sc.tl.umap(adata_merfish)
sc.tl.leiden(
adata_merfish,
key_added=”clusters”,
resolution=0.5,
n_iterations=2,
flavor=”igraph”,
directed=False,
)
adata_merfish
sc.pl.umap(adata_merfish, color=”clusters”)
sc.pl.embedding(adata_merfish, basis=”spatial”, color=”clusters”)

小结
好啦,今天的分享结束啦!Scanpy是与seurat的分析流程基本一致,都是质控-选择高变基因-PCA降维-聚类-UMAP(tSNE)降维这些步骤。但是,Python相比R有更好的内存管理,更丰富的人工智能算法资源,并且可以调用GPU加速计算。Python在图像识别、空间分割和去卷积等算法领域拥有更好的资源,因此很多优秀的空转分析工具都是python编写的。做好空转数据的分析不能局限于R语言生态,让我们从scanpy开始,拥抱python。最后小云给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!需要代码的也可以联系我呀。
