前言
天苍苍,野茫茫,一天不分享小果心痒痒!今天跟着小果来看看细胞通讯又有什么新花样了!自从单细胞爆火之后,在过去的几年中已经开发了好多种基于配体和受体表达的转录组数据中推断细胞间通讯网络的工具。像大家耳熟能详的cellchat等R包。很多的生信文章中都用到了这个包,是不是已经审美疲劳了?是不是想换换口味了?那就跟着小果来看看法国圣路易斯医院苏梅利斯教授团队的佳作吧!ICELLNET 配体-受体数据库从 380 个大幅扩展到 1669 个生物学策划的相互作用,整合了参与免疫串扰、细胞粘附和 Wnt 通路的重要通讯分子家族,优化了用于单细胞 RNA 测序数据分析的 ICELLNET 框架,提供了细胞间通讯结果的新可视化,以促进优先级排序和生物学解释。听到这里心动了吗!那就赶快跟着小果的步伐学起来!开始之前请允许小果打个广告,广告之后更精彩哦!练了十年生信分析的小果对于生信分析知识已经如鱼得水,相信有小果的加持一定能让大家学会!没有时间学习且有生信分析的小伙伴们快来滴滴小果噢!从分析到可视化直到你满意为止!

小果在这里给大家送福利了,有需要服务器的小伙伴们,欢迎大家联系小果,保证服务器的性价比最高哦!
教程
剖析癌症相关成纤维细胞亚群的细胞间通讯。癌症相关成纤维细胞 (CAF) 是位于肿瘤微环境中的基质细胞,已知可增强肿瘤表型,例如癌细胞增殖和炎症。先前的研究表明 CAFs 表型存在异质性。他们确定了 CAF 的 4 个亚群,包括 CAF-S1 和 CAF-S4 在三阴性乳腺癌 (TNBC) 中积累。CAF-S1 与免疫抑制微环境显著相关。
在本教程中,我们希望使用 TNBC 中 CAF-S1 和 CAF-S4 的可用转录谱来研究 CAF-S1 和 CAF-S4 与肿瘤微环境 (TME) 其他成分的通讯差异
1.相关R包安装
ICELLNET包是一个基于转录组学的框架,用于以全局方式剖析细胞通讯。它集成了一个由专家策划的原始配体-受体相互作用数据库,同时考虑了多个亚基表达。基于转录组图谱,ICELLNET软件包允许计算细胞之间的通信分数,并提供多种可视化模式,有助于深入研究细胞间相互作用机制并扩展生物学知识。
#下载并加载所有依赖项后,您可以加载“icellnet”包。
install.packages(c(“devtools”, “jetset”, “readxl”, “psych”, “GGally”, “gplots”, “ggplot2”, “RColorBrewer”, “data.table”, “grid”, “gridExtra”, “ggthemes”, “scales”,”rlist”)) ##Installs devtools and the icellnet CRAN dependancies
if (!requireNamespace(“BiocManager”, quietly = TRUE)) # Installs icellnet Bioconductor dependancies
install.packages(“BiocManager”)
BiocManager::install(c(“BiocGenerics”, “org.Hs.eg.db”, “hgu133plus2.db”, “annotate”))
library(devtools)
install_github(“soumelis-lab/ICELLNET”,ref=”master”, subdir=”icellnet”)
2..加载 ICELLNET 数据库并将数据库限制为不同的细胞因子家族
library(BiocGenerics)
library(“org.Hs.eg.db”)
library(“hgu133plus2.db”)
library(jetset)
library(ggplot2)
library(dplyr)
library(icellnet)
library(gridExtra)
##读取数据
db=as.data.frame(read.csv(curl::curl(url=”https://raw.githubusercontent.com/soumelis-lab/ICELLNET/master/data/ICELLNETdb.tsv”), sep=”\t”,header = T, check.names=FALSE, stringsAsFactors = FALSE, na.strings = “”))
my.selection.LR=c(“Cytokine”)
db2 <- db[grepl(paste(my.selection.LR, collapse=”|”),db$Family),] #if you want to use all the database, do instead : db2=db
db.name.couple=name.lr.couple(db2, type=”Subfamily”)
head(db.name.couple)
3. 从人类原代细胞图谱数据集中加载伴侣细胞类型
#从github下载PC.data.all和PC.target.all对象,并在你的Rstudio会话中打开它们
PC.data.all=as.data.frame(read.csv(“~/Downloads/PC.data.all.csv”, sep=”,”, header = T, check.names=FALSE, stringsAsFactors = FALSE, na.strings = “”))
rownames(PC.data.all)=PC.data.all$ID
PC.target.all=as.data.frame(read.csv(“~/Downloads/PC.target.all.csv”, sep=”,”,header = T, check.names=FALSE, stringsAsFactors = FALSE, na.strings = “”))
my.selection=c(“Epith”, “Fblast_B”, “Endoth”,”Mono”, “Macroph”, “pDC”, “DC2”, “DC1”, “NK”, “Neutrop”,”CD4 T cell”,”CD8 T cell”, “Treg”,”B cell”)
PC.target = PC.target.all[which(PC.target.all$Class%in%my.selection | PC.target.all$Class%in%my.selection),c(“ID”,”Class”,”Cell_type”)]
PC.data = PC.data.all[,PC.target$ID]
使用人类原代细胞图谱数据集作为伴侣细胞的后续步骤:
- 使用 hgu133plus2.db() 函数在 AffyID 和数据库中使用的基因符号之间创建转换图表。
- 执行 gene.scaling() 函数,这将 a) 选择与数据库中包含的配体和/或受体相对应的基因 (db)。b) 在 0 到 10 的所有条件下对每个配体/受体基因表达进行缩放。对于每个基因,最大值定义为“n”个最高表达值的平均表达量。将基因表达除以最大基因表达,然后乘以 10,将每个基因的数据表达矩阵分别缩放在 0 到 10 之间。 n 的默认值为 1。 在此示例中,将 n 设置为 18,以便将 5% 极值表达值的平均值作为伴侣细胞的最大值。
### 将基因符号转换为ID
PC.affy.probes = as.data.frame(PC.data[,c(1,2)])
PC.affy.probes$ID = rownames(PC.affy.probes) # for format purpose
transform = db.hgu133plus2(db2,PC.affy.probes) # creation of a new db2 database with AffyID instead of gene symbol
##伴侣细胞数据集的基因缩放
PC.data=gene.scaling(data = PC.data, n=18, db = transform)
4. CAF-S1 和 CAF-S4 转录组学谱
该数据集提供了 77 个样本的基因表达矩阵,这些样本对应于 Costa 等人于 2018 年生成的 CAF 亚群转录组图谱,用于识别乳腺癌中的四个 CAF 亚群。
#中心细胞数据文件(处理过的基因表达矩阵)
data=as.data.frame(read.table(curl::curl(url=”https://raw.githubusercontent.com/soumelis-lab/ICELLNET/master/data_CAF/data_CAF.txt”), sep=”\t”, header = T))
rownames(data)=data$SYMBOL
data=dplyr::select(data, -SYMBOL)
CC.data= gene.scaling(data = data, n=4, db = db2)
#目标中心细胞文件(不同样本的描述)
CC.target = as.data.frame(read.table(curl::curl(url=”https://raw.githubusercontent.com/soumelis-lab/ICELLNET/master/data_CAF/target_CAF.txt”),sep = “\t”,header=T))
head(CC.target)
与 PC.data 相同,将基因表达矩阵重新调整为 0 到 10,将所有 CAF 样本考虑到尽可能大的分布。 在这个阶段,我们选择感兴趣的样本,这意味着在肿瘤(TNBC癌症类型)中发现的CAF-S1或CAF-S4。这相当于 CAF-S1 的 6 个样品和 CAF-S4 的 3 个样品。
CC.selection.S1 = CC.target[which(CC.target$Type==”T”&CC.target$subset==”S1″&CC.target$Cancer.subtype==”TN”),”Sample.Name”] # CAF-S1 in TNBC samples
CC.selection.S2 = CC.target[which(CC.target$Type==”T”&CC.target$subset==”S4″&CC.target$Cancer.subtype==”TN”),”Sample.Name”] # CAF-S4 in TNBC samples
CC.data.selection.S1 = CC.data[,which(colnames(CC.data)%in%CC.selection.S1)]
CC.data.selection.S2 = CC.data[,which(colnames(CC.data)%in%CC.selection.S2)]
5. ICELLNET细胞间通讯分数的计算
细胞间通讯的定量包括对具有已知表达谱的两种细胞类型之间每种配体/受体相互作用的强度进行评分。不对 L/R 表达式应用筛选阈值。如果通讯分子(配体或受体或两者兼而有之)未由细胞表达,则分数将为零。默认情况下,数据库的所有交互都被视为计算分数。也可以通过手动选择数据库中的特定分子家族或考虑DEG来计算分数来减少相互作用的数量,具体取决于生物学问题。
在这里,我们计算向外通信分数(direction = “out”),这意味着从 CAF 子集到伙伴细胞的通信。 这意味着我们考虑CAF表达的配体和伴侣细胞表达的受体来计算细胞间通讯分数。
score.computation.1= icellnet.score(direction=”out”, PC.data=PC.data, CC.data= CC.data.selection.S1,
PC.target = PC.target, PC=my.selection, CC.type = “RNAseq”,
PC.type = “Microarray”, db = db2)
score1=as.data.frame(score.computation.1[[1]])
lr1=score.computation.1[[2]]
score.computation.2= icellnet.score(direction=”out”, PC.data=PC.data, CC.data= CC.data.selection.S2,
PC.target = PC.target,PC=my.selection, CC.type = “RNAseq”,
PC.type = “Microarray”, db = db2)
score2=as.data.frame(score.computation.2[[1]])
lr2=score.computation.2[[2]]
Scores=cbind(score1,score2)
colnames(Scores)=c(“CAF-S1″,”CAF-S4”)
Scores
score1 和 score2 对应于全局分数,即各个分数的总和。分数矩阵 (Scores) 对应于 ICELLNET 在所有外围小区和中心小区之间计算的全局通信分数的摘要。LR1 和 LR2 对应于单个分数矩阵,对于进一步的可视化步骤很有用。
6. 使用不同的可视化模式深入研究细胞间通信
6.1细胞间通讯网络表示
这允许通过network.create()功能以全局方式可视化细胞间通信网络。在这些有向图中,节点表示像元类型,连接两种像元类型的边的宽度与它们之间通信强度的全局测量成正比,箭头表示通信方向。
#显示通信网络
PC.col = c(“Epith”=”#C37B90”, “Muscle_cell”=”#c100b9″,”Fblast_B”=”#88b04b”, “Fblast”=”#88b04b”,”Endoth”=”#88b04b”,
“Mono”=”#ff962c”,”Macroph”=”#ff962c”,”moDC”=”#ff962c”,”DC1″=”#ff962c”,”DC2″=”#ff962c”,”pDC”=”#ff962c”,”NK”=”#ff962c”,”Neutrop”=”#ff962c”,
“CD4 T cell”=”#5EA9C3″,”CD8 T cell”=”#5EA9C3″,”Treg”=”#5EA9C3″,”B cell”=”#5EA9C3”)
network.plot1 = network.create(icn.score = Scores[1], scale = c(round(min(Scores)-1),round(max(Scores))+1), direction = “out”, PC.col)
network.plot2 = network.create(icn.score =Scores[2], scale = c(round(min(Scores)-1),round(max(Scores))+1), direction = “out”,PC.col)
gridExtra::grid.arrange(network.plot1, network.plot2, ncol=2, nrow=1)

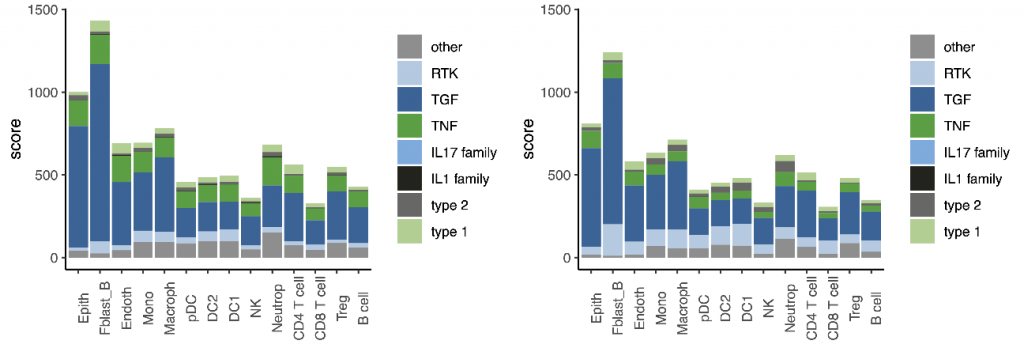
6.2通讯分子分布 – 条形图表示
LR.family.score()允许在分子类别的水平上剖析全局分数,并允许识别来自同一家族的共表达分子的模式。这一层分析有助于定性层面的解释。 每个分子家族对通信分数的贡献可以显示为热图(设置 plot=“heatmap”时)或条形图(设置 plot=“barplot”时)。
##标签和范围定义
my.family=c(“type 1”, “type 2”, “IL1 family”, “IL17 family”, “TNF”,”TGF”,”RTK”)
family.col = c( “type 1″= “#A8CF90”, “type 2″= “#676766”, “IL1 family”= “#1D1D18” ,
“IL17 family” =”#66ABDF”, “TNF” =”#12A039″, “TGF” = “#156399”, “RTK”=”#AECBE3”, “other” = “#908F90″,”NA”=”#908F90”)
ymax=round(max(Scores))+1 #to define the y axis range of the barplot
# to get family contribution as dataframe
LR.family.score(lr=lr1, family=my.family, db.couple=db.name.couple, plot= NULL)
# 条形图
contrib.family1= LR.family.score(lr=lr1, family=my.family, db.couple=db.name.couple, plot=”barplot”, ymax=ymax, family.col=family.col)
contrib.family2= LR.family.score(lr=lr2, family=my.family, db.couple=db.name.couple, plot=”barplot”, ymax=ymax, family.col=family.col)
gridExtra::grid.arrange(contrib.family1, contrib.family2, ncol=2, nrow=1)
6.3通信分数分布 – 气泡图表示
lr_ind=cbind(lr1[,”Fblast_B”],lr2[,”Fblast_B”])
colnames(lr_ind)=c(“S1_Fblast”, “S4_Fblast”)
balloon=icellnet::LR.balloon.plot(lr = lr_ind, sort.by=”var”, thresh = 25, topn=15, db.name.couple=db.name.couple, title=”Fblast”, family.col=family.col)
balloon

6.4 P值计算以比较通信分数
可以计算两种类型的p值(函数),以比较从同一中心细胞获得的通信分数与不同的伙伴细胞(between=“cells”),或比较从对应于不同生物条件的两个不同中心细胞获得的通信分数与同一伙伴细胞(between=“conditions”)。如果 between=“cells”,则考虑中心细胞配体的平均表达来计算通讯分数,并且每个配体分别复制伴侣细胞的受体表达。通过这种方式,对于一个伴侣细胞,我们获得了 n 个通信分数的分布,即该特定细胞类型复制的伴侣细胞数量。如果 between=“conditions”,则计算通信分数时会考虑中心细胞的每次重复,以及伴侣细胞的平均基因表达。我们获得了 n 个通信分数的分布,n 个单位是在一个生物条件下中心细胞重复的数量。然后,执行 Wilcoxon 统计检验以比较通信分数分布。
# 从CAF-S1和不同的伴侣细胞获得的通信分数的比较
pvalue1=icellnet.score.pvalue(direction=”out”, PC.data=PC.data, CC.data= CC.data.selection.S1,
PC.target = PC.target,PC=my.selection, CC.type = “RNAseq”,PC.type = “Microarray”,
db = db2, between=”cells”, method=”BH”)[[1]]
pvalue.plot1=pvalue.plot(pvalue1, PC=my.selection)
pvalue.plot1

小结
ICELLNET是一种通用方法,可应用于广泛的转录组学数据,包括批量RNAseq、scRNAseq和空间转录组学。ICELLNET配体-受体数据库的重大富集将覆盖更多的生物学领域,这将扩展其性能并拓宽其应用领域。新的图形可视化对于促进结果的优先排序和解释至关重要,并将提供新的生物学见解。最后小果给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!
