前言
太无语了!最近很多同学向小果抱怨,刚刚学会怎么使用CellRank进行细胞轨迹分析,结果又更新了!小果为了安抚大家的情绪,赶紧撇下自己的实验,马不停蹄的给大家奉上一份使用教程!现有的轨迹推断方法主要设计用于快照单细胞RNA测序数据,无法容纳额外信息,如实验时间点、多模态测量、RNA速度和代谢标记。CellRank 2是一个多功能、可扩展的框架,用于研究多达数百万细胞的多视图单细胞数据中的细胞命运。通过分离转换矩阵的推断和分析,CellRank 2能够以一致和统一的方式克服单数据类型方法的局限性。下面赶快跟着小果来学习一下吧!没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找小果哦!练了十年生信分析的小果对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!
CellRank2的主要应用:
- 根据不同数量的生物学先验(包括伪时间、发育潜力、RNA 速度)
- 计算初始、终端和中间宏观状态
- 推断命运概率和驱动基因
- 可视化和聚类基因表达趋势

单细胞数据处理起来占用内存实在太大了,放过自己的电脑吧!小果在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系小果,保证服务器的性价比最高哦!
代码教程
1. 导入包和数据
import sys
if “google.colab” in sys.modules:
!pip install -q git+https://github.com/theislab/cellrank
import cellrank as cr
import scanpy as sc
sc.settings.set_figure_params(frameon=False, dpi=100)
cr.settings.verbosity = 2
import warnings
warnings.simplefilter(“ignore”, category=UserWarning)
##我们将使用人类骨髓的scRNA-seq数据集
##数据获取地址:https://data.humancellatlas.org/explore/projects/091cf39b-01bc-42e5-9437-f419a66c8a45
adata = cr.datasets.bone_marrow()
adata
## 将MAGIC插补数据移动到 layers
adata = adata[:, adata.var[“palantir”]].copy()
adata.layers[“MAGIC_imputed_data”] = adata.obsm[“MAGIC_imputed_data”].copy()
2. 预处理数据
#过滤和规范化
sc.pp.filter_genes(adata, min_cells=5)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, n_top_genes=3000)
#计算 PCA 和 k 最近邻 (k-NN) 图。
sc.tl.pca(adata, random_state=0)
sc.pp.neighbors(adata, random_state=0)
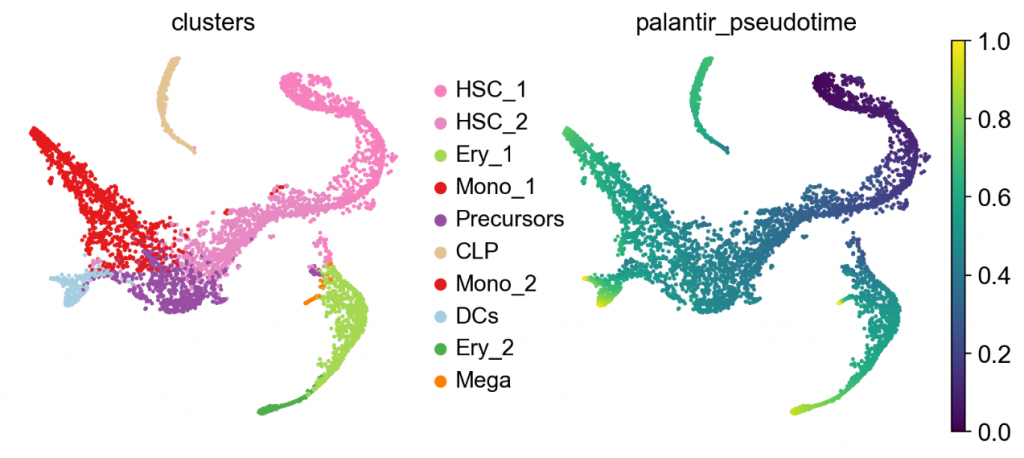
#使用原始研究提供的 t-SNE 嵌入、聚类标签和 Palantir 伪时间可视化这些数据
sc.pl.embedding(adata, basis=”tsne”, color=[“clusters”, “palantir_pseudotime”])
3. 使用内核
为了构造一个过渡矩阵,CellRank 在 中提供了许多核类。为了演示这个概念,我们将使用 k-NN 图边缘来指向增加伪时间的方向
from cellrank.kernels import PseudotimeKernel
pk = PseudotimeKernel(adata, time_key=”palantir_pseudotime”)
pk
请注意,内核需要一个对象来从中读取数据 – CellRank 是 Scanpy/AnnData 生态系统的一部分。唯一的例外是它直接接受转换矩阵,从而可以从 scanpy/AnnData 世界外部连接到 CellRank。
pk.compute_transition_matrix()
pk
为了获得该数据集中细胞动力学的第一印象,我们可以从造血干细胞 (HSC) 开始,在过渡基质隐含的马尔可夫链上进行模拟,并在 t-SNE 嵌入中可视化这些细胞。
pk.plot_random_walks(
seed=0,
n_sims=100,
start_ixs={“clusters”: “HSC_1”},
basis=”tsne”,
legend_loc=”right”,
dpi=150,
)
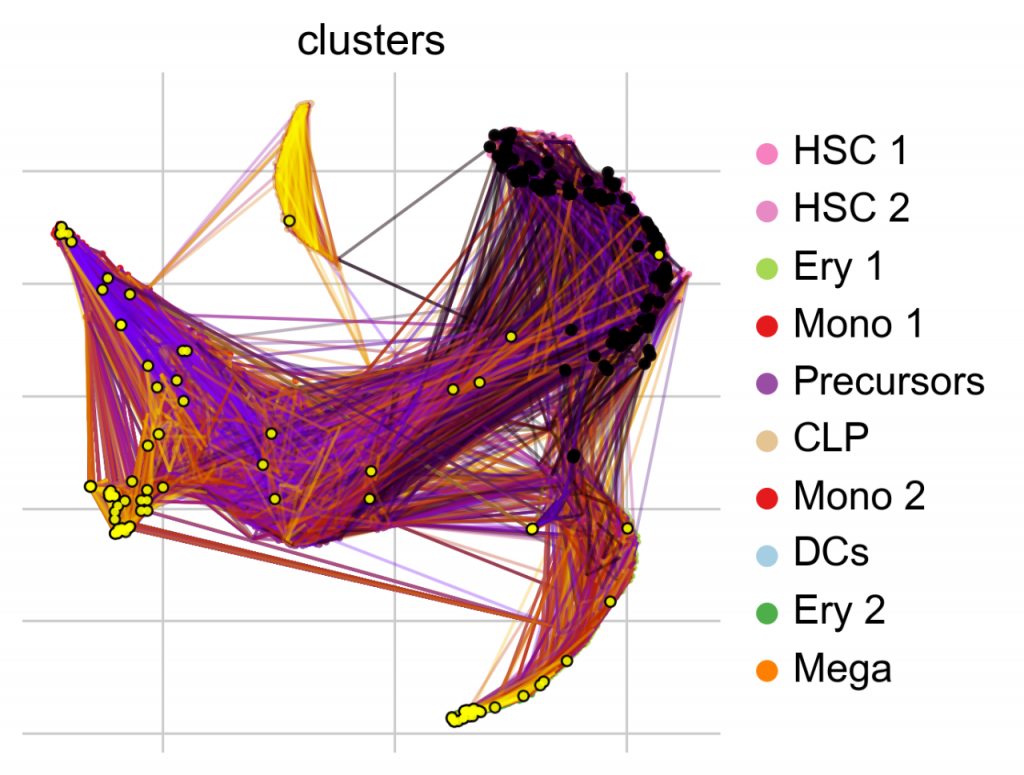
黑点和黄点分别表示随机行走开始和终端细胞。总体而言,这反映了人类造血中已知的分化层次。
4. 内核概述
每种模式和生物系统都有其自身的优势和局限性,因此仔细选择内核很重要。我们在下图中提供了一些指导。
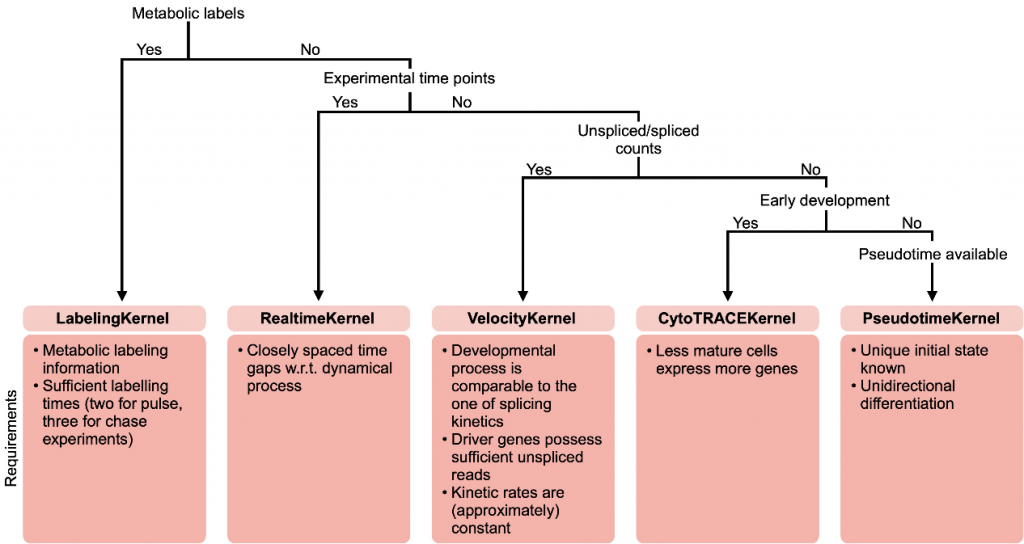
内核选择指南:CellRank内核支持不同的数据模式和生物系统。每个都有自己的假设和局限性。
如果您的数据已经有一个细胞-细胞转换矩阵,则可以使用 CellRank 的接口与外部方法直接传递该矩阵。
任何两个内核都可以通过加权平均值进行全局组合。为了证明这一点,让我们根据基因表达相似性设置一个
from cellrank.kernels import ConnectivityKernel
ck = ConnectivityKernel(adata).compute_transition_matrix()
ck
##与上面的伪时间内核结合使用
combined_kernel = 0.8 * pk + 0.2 * ck
combined_kernel
5. 写入和读取内核
有时,我们可能希望将内核写入磁盘,以便稍后继续分析,或者将其传递给其他人。这很容易做到;只需将内核写入 adata,然后将 adata 写入 file:
pk.write_to_adata()
adata.write(“hematopoiesis.h5ad”)
#为了继续分析,我们从磁盘读取对象,并从 AnnData 对象初始化一个新内核
adata = sc.read(“hematopoiesis.h5ad”)
pk_new = cr.kernels.PseudotimeKernel.from_adata(adata, key=”T_fwd”)
pk_new
6. 设置估算器
from cellrank.estimators import GPCCA
g = GPCCA(pk_new)
print(g)
7. 识别初始和结束状态
g.fit(n_states=10, cluster_key=”clusters”)
g.plot_macrostates(which=”all”)
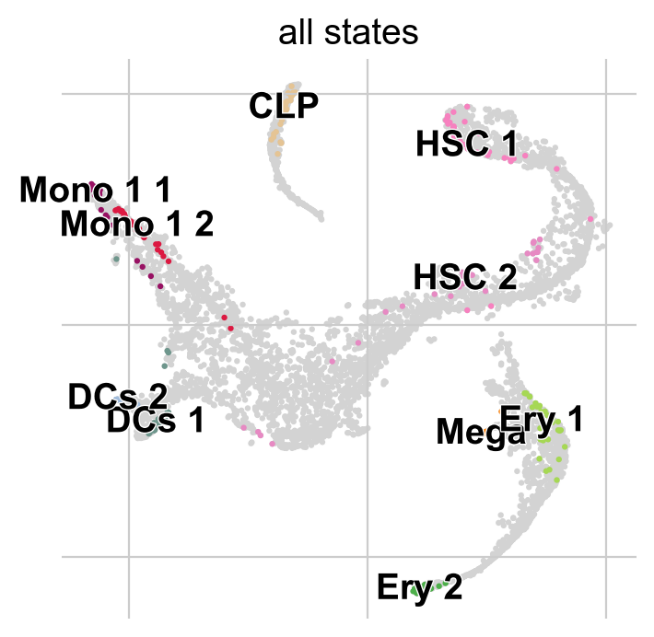
#对于每个宏状态,该算法会计算关联的稳定性值。
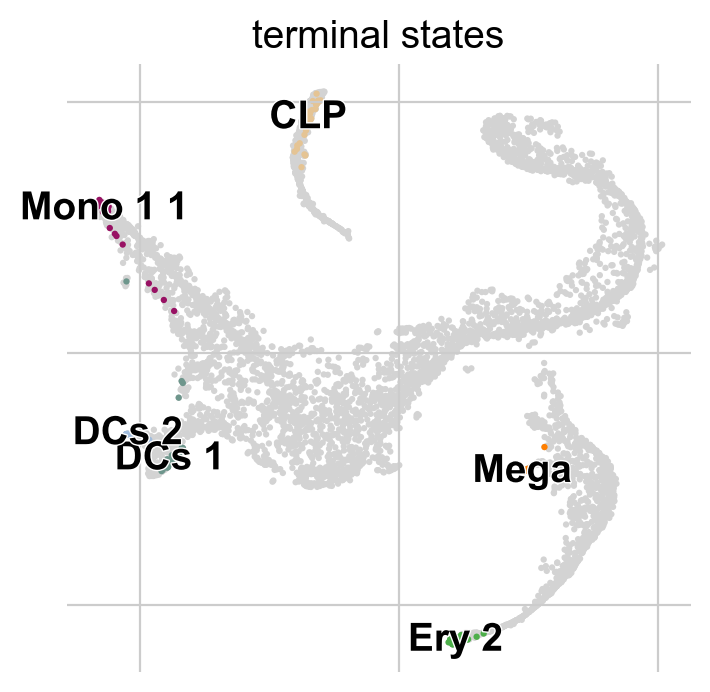
g.predict_terminal_states(method=”top_n”, n_states=6)
g.plot_macrostates(which=”terminal”)
8. 计算命运概率和驱动基因
#我们现在可以计算每个细胞达到每个终端状态的可能性。
g.compute_fate_probabilities()
g.plot_fate_probabilities(legend_loc=”right”)
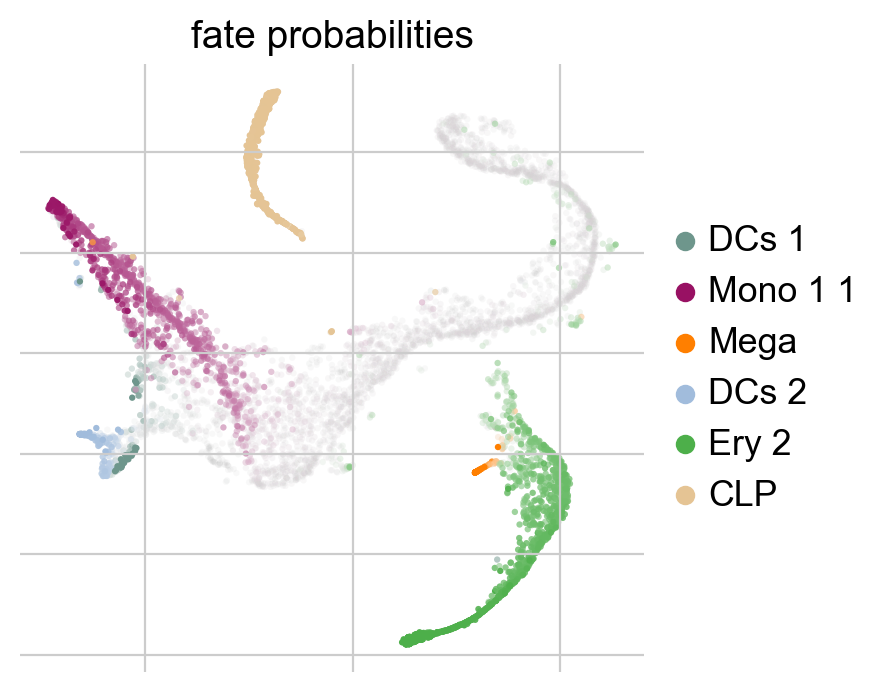
上图结合了所有终末状态的命运概率,每个细胞根据其最可能的命运着色;颜色强度反映了谱系启动的程度。
cr.pl.circular_projection(adata, keys=”clusters”, legend_loc=”right”)

每个点代表一个细胞,由簇标签着色。细胞根据其命运概率排列在圆圈内;命运偏向的细胞被放置在它们相应的角落旁边,而幼稚的细胞被放置在中间。
##为了推断这些轨迹中的任何一个的假定驱动基因,我们将表达值与命运概率相关联。
mono_drivers = g.compute_lineage_drivers(lineages=”Mono_1_1″)
mono_drivers.head(10)
9. 可视化表达趋势
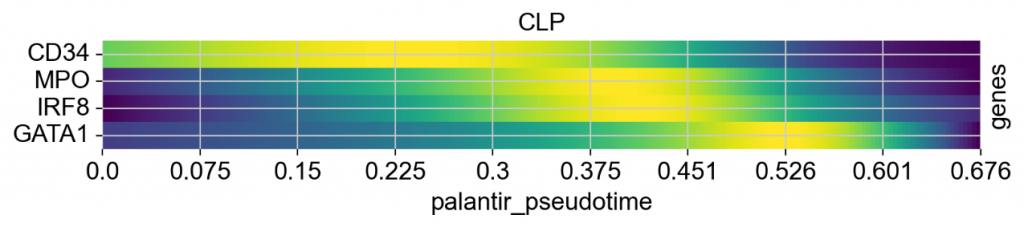
给定命运概率和伪时间,我们可以绘制轨迹特异性基因表达趋势。具体来说,我们拟合广义加法模型(GAM),根据其命运概率向量对每个细胞的贡献进行加权。
model = cr.models.GAMR(adata)
##初始化模型后,我们可以沿着特定轨迹可视化基因动力学。
cr.pl.gene_trends(
adata,
model=model,
data_key=”MAGIC_imputed_data”,
genes=[“GATA1”, “CD34”, “IRF8”, “MPO”],
same_plot=True,
ncols=2,
time_key=”palantir_pseudotime”,
hide_cells=True,
)

在上面,我们按基因对表达趋势进行了分组,并可视化了每个面板的几个轨迹。我们也可以反过来做 – 按轨迹对表达趋势进行分组,每个面板可视化几个基因。
cr.pl.heatmap(
adata,
model=model,
data_key=”MAGIC_imputed_data”,
genes=[“GATA1”, “CD34”, “IRF8”, “MPO”],
lineages=[“Mono_1_1”, “Mega”, “CLP”],
time_key=”palantir_pseudotime”,
cbar=False,
show_all_genes=True,
)


小结
总之,CellRank 2是一个健壮、模块化且可扩展的框架,能够在多视图单细胞数据中统一研究细胞命运。它不仅克服了现有方法的局限性,还展示了其在解析复杂细胞状态变化和分化轨迹中的卓越性能。研究团队期待未来的工作能够进一步扩展CellRank 2,以适应更多新兴数据模态,并结合干扰数据和因果推断,深入理解分子驱动因素,为科学研究提供更强大的工具。最后小果给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!
