前言
大海哥在群里看到小伙伴们对于生信分析到底还能不能发SCI讨论的热火朝天!很多小伙伴们@大海哥,想听听大海哥的想法!传统的老套路想发SCI实在是太难了。怎样才能脱离生信内卷赛道发表SCI呢?对于大多数的临床医生来说,生信分析很难再算法上进行创,那还有什么办法呢?那就是卷工作量、卷新的分析方法!今天大海哥带着大家来看看单细胞测序分析还能怎么卷?Mellon带你了解单细胞分化过程中的细胞密度变化,为指导生物轨迹的机制提供见解的潜力。小伙伴们快来学习一下,看看如何应用到自己的分析中充实工作量呢!
练了十年生信分析的大海哥对于生信分析知识已经如鱼得水,相信有大海哥的加持一定能让大家学会!没有时间学习且有生信分析的小伙伴们快来滴滴大海哥噢!从分析到可视化直到你满意为止!
大海哥在这里给大家送福利了,有需要服务器的小伙伴们,欢迎大家后台私信大海哥哦,保证服务器的性价比最高哦!
代码教程
本教程提供了在单细胞 RNA 测序 (scRNA-seq) 数据上应用Mellon密度估计器的指南。本教程的目的是说明使用 Mellon 软件包执行密度估计所需的步骤。
在开始之前,请确保安装了以下必备软件包,这些软件包对于将 Mellon 与 scRNA-seq 数据结合使用至关重要:
- scanpy: https://scanpy.readthedocs.io/en/stable/installation.html(大海哥已经带着大家学习过了哦)
- palantir: https://github.com/dpeerlab/Palantir.我们建议使用pip install git+https://github.com/dpeerlab/Palantir
1.加载相应的包
import numpy as np
import matplotlib
import matplotlib.pyplot as plt
from sklearn.cluster import k_means
import palantir
import mellon
import scanpy as sc
import warnings
from numba.core.errors import NumbaDeprecationWarning
warnings.simplefilter(“ignore”, category=NumbaDeprecationWarning)
%matplotlib inline
matplotlib.rcParams[“figure.figsize”] = [4, 4]
matplotlib.rcParams[“figure.dpi”] = 125
matplotlib.rcParams[“image.cmap”] = “Spectral_r”
# no bounding boxes or axis:
matplotlib.rcParams[“axes.spines.bottom”] = “on”
matplotlib.rcParams[“axes.spines.top”] = “off”
matplotlib.rcParams[“axes.spines.left”] = “on”
matplotlib.rcParams[“axes.spines.right”] = “off”
2. 读取和显示数据集
我们将从加载 scRNA-seq 数据集开始。我们将使用预处理的 T 细胞耗竭骨髓数据。
ad_url = “https://fh-pi-setty-m-eco-public.s3.amazonaws.com/mellon-tutorial/preprocessed_t-cell-depleted-bm-rna.h5ad”
ad = sc.read(“data/preprocessed_t-cell-depleted-bm-rna.h5ad”, backup_url=ad_url)
ad
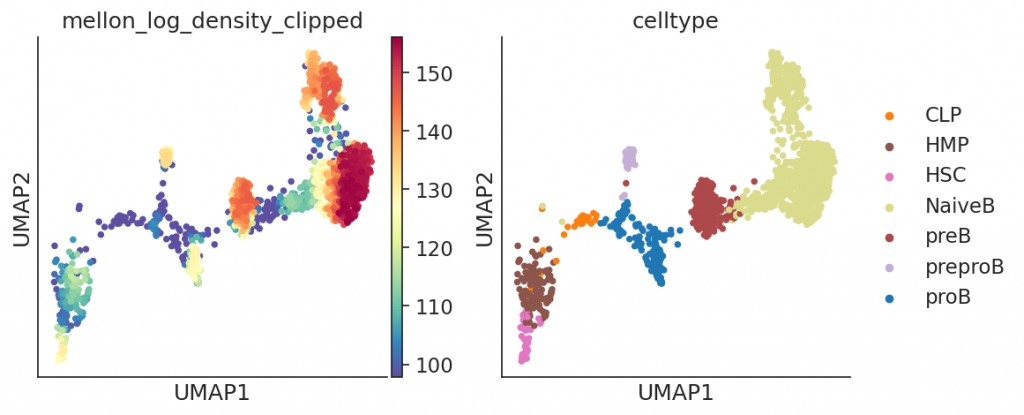
sc.pl.scatter(ad, basis=”umap”, color=”celltype”)
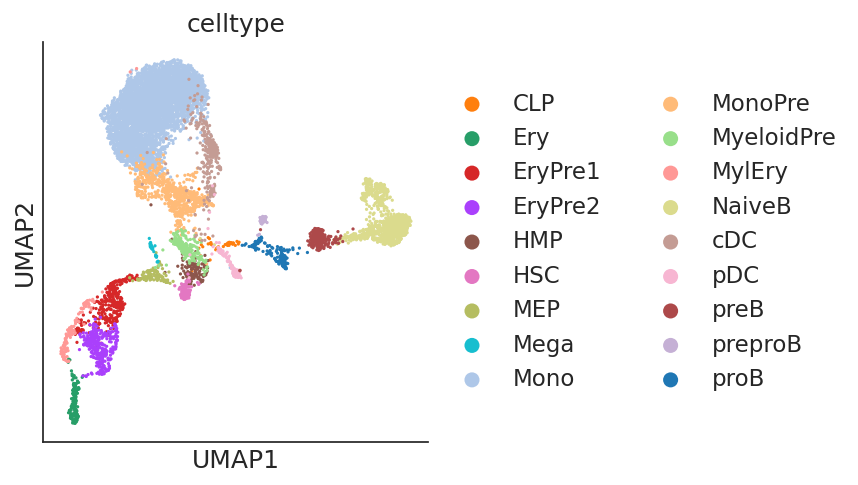
3. 预处理
我们将使用扩散图作为细胞状态表示的输入作为 Mellon 的输入。扩散图通过创建保持细胞之间关系的降维表示来捕获数据的固有几何形状。可以使用该函数计算扩散图。默认情况下,此函数使用预先计算的函数,但可以更改为使用 anndata 对象中的任何条目。
%%time
dm_res = palantir.utils.run_diffusion_maps(ad, pca_key=”X_pca”, n_components=20)
4. 密度计算
在此步骤中,我们将使用 Mellon 的 DensityEstimator 类计算细胞状态密度。上面计算的扩散分量用作输入。可以使用UMAP可视化计算密度。
%%time
model = mellon.DensityEstimator()
log_density = model.fit_predict(ad.obsm[“DM_EigenVectors”])
predictor = model.predict
ad.obs[“mellon_log_density”] = log_density
ad.obs[“mellon_log_density_clipped”] = np.clip(
log_density, *np.quantile(log_density, [0.05, 1])
)
ad
sc.pl.scatter(
ad, color=[“mellon_log_density”, “mellon_log_density_clipped”], basis=”umap”
)
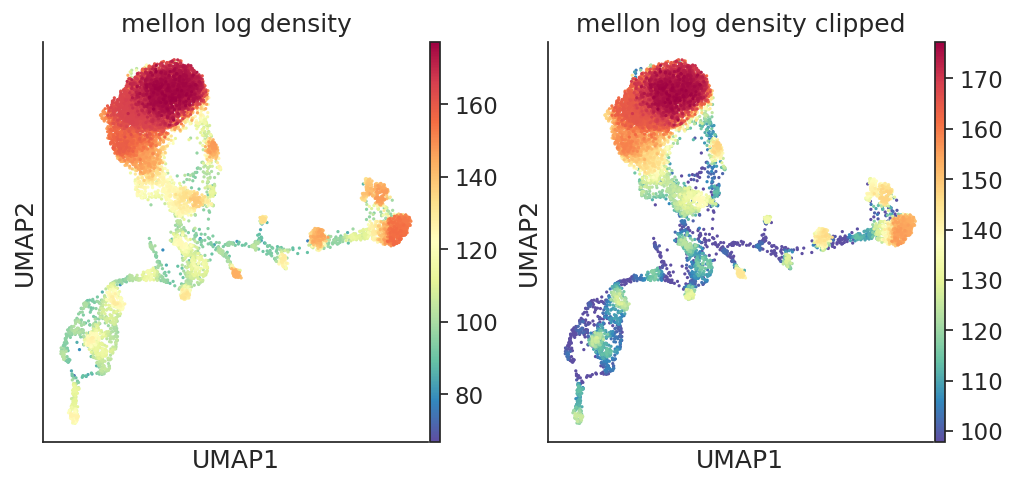
5.分析细胞类型
接下来,我们可以使用计算出的细胞密度来分析数据集中的不同细胞类型。
fig, (ax1, ax2) = plt.subplots(1, 2, width_ratios=[3, 2], figsize=[12, 4])
sc.pl.violin(ad, “mellon_log_density”, “celltype”, rotation=45, ax=ax1, show=False)
sc.pl.scatter(ad, color=”celltype”, basis=”umap”, ax=ax2, show=False)
plt.show()
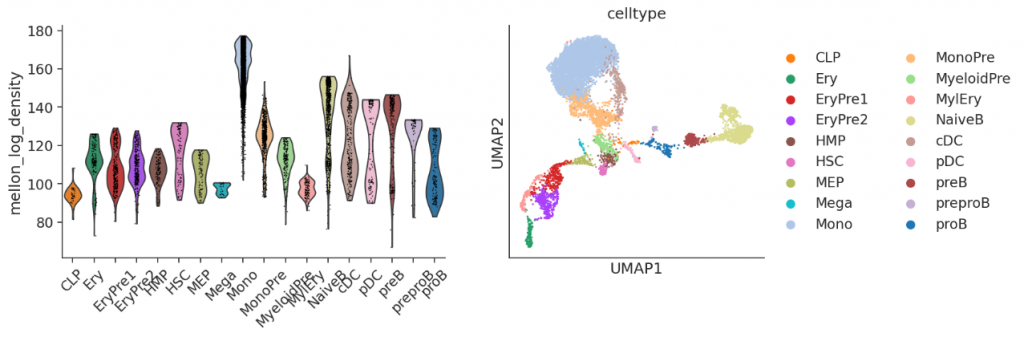
#沿伪时间的密度比较
##密度可以与伪时间轴或其他轴进行比较,以识别高密度和低密度区域。
ad.obs[“palantir_pseudotime”], ad.obsm[“palantir_lineage_cells”]
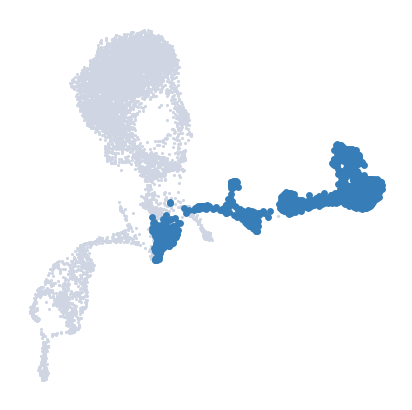
##我们将探索沿 B 细胞谱系的密度
bcell_lineage_cells = ad.obs_names[ad.obsm[“palantir_lineage_cells”][“NaiveB”]]
palantir.plot.highlight_cells_on_umap(ad, bcell_lineage_cells)
plt.show()
import pandas as pd
ct_colors = pd.Series(
ad.uns[“celltype_colors”], index=ad.obs[“celltype”].values.categories
)
plt.figure(figsize=[8, 3])
plt.scatter(
ad.obs[“palantir_pseudotime”][bcell_lineage_cells],
ad.obs[“mellon_log_density”][bcell_lineage_cells],
s=10,
color=ct_colors[ad.obs[“celltype”][bcell_lineage_cells]],
)
plt.show()
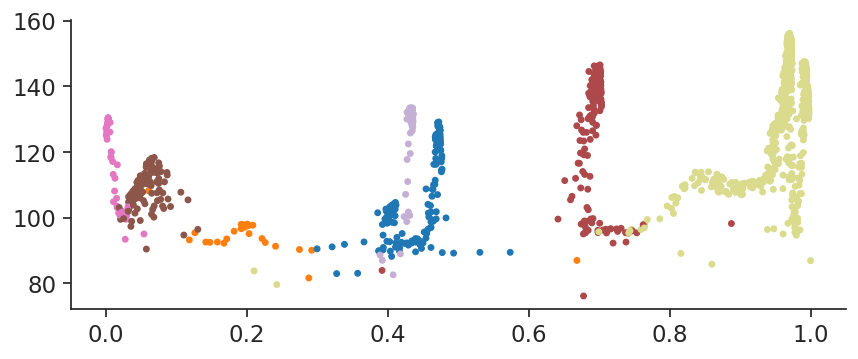
palantir.plot.plot_branch(
ad,
branch_name=”NaiveB”,
position=”mellon_log_density”,
color=”celltype”,
masks_key=”palantir_lineage_cells”,
s=100,
)
plt.show()

该图清楚地说明了定义 B 细胞分化的交替高密度和低密度区域。可以检查这种变异性以识别高密度和低密度区域.
##相同的功能可用于可视化基因表达
# 在伪时间和对数密度的比较中绘制EBF1的估算表达
palantir.plot.plot_branch(
ad,
branch_name=”NaiveB”,
position=”mellon_log_density”,
color=”EBF1″,
color_layer=”MAGIC_imputed_data”,
masks_key=”palantir_lineage_cells”,
s=100,
)
plt.show()

6表达的局部变异性或局部变化
局部变异性提供了每个细胞状态的基因表达变化的度量。这是通过将细胞中的基因与其相邻细胞状态进行比较来确定的,并且可以使用以下方法计算:palantir.utils.run_local_variability
# 基因的局部变化
palantir.utils.run_local_variability(ad)
#这将添加’ local_variability ‘作为一个图层到anndata
ad
# 局部可变性或局部变化也可以使用’ plot_branch ‘函数进行可视化
palantir.plot.plot_branch(
ad,
branch_name=”NaiveB”,
position=”mellon_log_density”,
color=”EBF1″,
color_layer=”local_variability”,
masks_key=”palantir_lineage_cells”,
s=100,
)
plt.show()

#使用蓝色色图来绘制变化
palantir.plot.plot_branch(
ad,
branch_name=”NaiveB”,
position=”mellon_log_density”,
color=”EBF1″,
color_layer=”local_variability”,
masks_key=”palantir_lineage_cells”,
s=100,
cmap=matplotlib.cm.Blues,
edgecolor=”black”,
linewidth=0.1,
)
plt.show()

# umap可以与细胞子集一起绘制以探索密度
sc.pl.embedding(
ad[bcell_lineage_cells],
basis=”umap”,
color=[“mellon_log_density_clipped”, “celltype”],
)
7. 保存和加载变量
可以将预测变量序列化到字典中,并直接保存在 Anndata 对象中。然后,我们可以将 Anndata 对象写入磁盘,重新加载它,从保存的字典中重构预测器,并将其应用于我们的数据。最终检查验证反序列化密度函数是否与原始函数相同。
# 转换为字典并保存到Anndata,然后将Anndata写入磁盘
ad.uns[“log_density_function”] = predictor.to_dict()
ad.write(“data/adata.h5ad”)
#加载变量
#重新加载Anndata
ad = sc.read(“data/adata.h5ad”)
predictor = mellon.Predictor.from_dict(ad.uns[“log_density_function”])
# 验证
log_density = predictor(ad.obsm[“DM_EigenVectors”])
assert np.all(
np.isclose(log_density, ad.obs[“mellon_log_density”])
), “Deserialized density function differs from original.”
小结
细胞状态密度表征了细胞沿表型景观的分布,对于揭示驱动不同生物过程的机制至关重要。在这里,大海哥给小伙伴们介绍了Mellon,一种从单细胞数据的高维表示中估计细胞状态密度的算法。Mellon 有助于跨各种单细胞数据模式进行密度估计,并随细胞数量线性缩放,这在细胞状态密度在理解分化过程方面的重要性。最后大海哥给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!
