在复杂的生物网络中,如何精确地识别出那些调控疾病发生发展的关键基因途径呢?小师妹今天将引导您深入了解pathwayPCA这一强大的生物信息学分析工具,它能够揭示基因表达数据背后的生物学意义,帮助我们理解疾病的分子机制。
pathwayPCA集成了基于主成分分析(PCA)的途径分析方法,这些方法最初由Chen等人在2008年、2010年以及2011年的研究中提出。它不仅能够帮助我们测试基因途径与各种表型之间的关联,还能提取出那些在途径中起着关键作用的基因,计算出代表个体样本途径活动的主成分。更为神奇的是,它能够揭示影响途径显著性的关键基因及其数据,提高我们的分析效率,即使是最复杂的实验设计也能迎刃而解。
请注意,pathwayPCA在处理大型数据集时可能需要较大的内存,因此小师妹建议使用服务器进行分析。如果您需要性价比高的服务器租赁服务,欢迎联系小师妹。
接下来,小师妹将以卵巢癌数据作为案例,一步步指导您完成从数据预处理到执行途径分析的整个流程,确定与生存结果相关的蛋白表达的重要途径。我们将一起揭开基因表达数据的神秘面纱,探索其背后的生物学意义。如果在分析过程中遇到任何问题,小师妹随时准备为您提供帮助。
首先,我们需要安装pathwayPCA包。如果您尚未安装,可以通过以下命令进行安装:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(version=’devel’)
BiocManager::install(“pathwayPCA”)
安装完成后,我们可以通过以下命令加载pathwayPCA包:
library(tidyverse)
library(pathwayPCA)
- 数据准备
在本例中,我们将使用临床蛋白质组学肿瘤分析联盟(CPTAC)最近生成的基于质谱的卵巢癌的蛋白质组学数据。归一化蛋白丰度表达数据集可从LinkedOmics数据库http://linkedomics.org/data_download/TCGA-OV/获得。我们选用了由太平洋西北国家实验室(PNNL)生成的“Proteome (PNNL, Gene level)”数据集。
首先,我们需要创建一个存储1的omics类数据对象(包含表达式数据集、样本的表型信息、途径集合)。
- 表达和表型数据
我们可以通过加载ovarian_PNNL_survival获得卵巢癌数据集。
# 下载数据
gitHubPath_char <- “https://raw.githubusercontent.com/lizhongliu1996/pathwayPCAdata/master/”
ovSurv_df <- readRDS(
url(paste0(gitHubPath_char, “ovarian_PNNL_survival.RDS”))
)
# 或者直接加载提供的数据集
ovSurv_df <- read.csv(“ovSurv_df.csv”)
ovSurv_df [1:5, 1:5]

ovSurv_df数据集是一个包含蛋白质表达水平和与样本id匹配的存活结果的数据框架。变量(列)包括总体存活时间和审查状态,以及83个样品中每个5162个蛋白的表达数据。
- 途径集合
在进行pathway分析时,我们需要收集一系列的生物学途径。对于这些途径的收集,我们需要指定一个.gmt文件,这是一个文本文件,其中每一行对应一个途径。每一行包含一个ID(TERMS列)、一个可选的描述(description列),以及途径中的基因(所有后续列)。pathwayPCA包含了2018年6月的Wikipathways集合,适用于智人(Homo sapiens),可以使用read_gmt函数加载:
print(“wikipathways-20190110-gmt-Homo_sapiens.gmt”)
dataDir_path <- system.file(“extdata”, package = “pathwayPCA”, mustWork = TRUE)
wikipathways_PC <- read_gmt(paste0(dataDir_path, “/wikipathways_human_symbol.gmt”),description = TRUE)
- 创建OmicsSurv数据容器
现在我们已经准备好了三种数据(表达式数据集、样本的表型信息、途径集合),我们可以创建一个OmicsSurv数据容器。
要查看我们刚刚创建的Omics数据对象的摘要,只需输入对象的名称即可。
ov_OmicsSurv <- CreateOmics(
# protein expression data
assayData_df = ovSurv_df[, -(2:3)],
# pathway collection
pathwayCollection_ls = wikipathways_PC,
# survival phenotypes
response = ovSurv_df[, 1:3],
# phenotype is survival data
respType = “survival”,
# retain pathways with > 5 proteins
minPathSize = 5
)
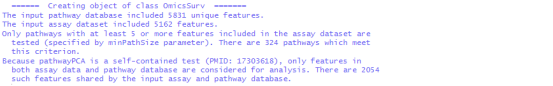
我们可以查看刚刚这个Omics数据对象的摘要:

- 测试途径与表型的相关性
- 方法描述
创建Omics对象后,我们可以采用两种方法分析数据:AES-PCA和SuperPCA。AES-PCA是一种无监督方法,通过降维技术提取每个途径的主成分,并用样本标签的随机置换来测试这些成分与表型的关系。而SuperPCA则是一种监督方法,它选择与疾病结果密切相关的基因子集来估计途径的潜在变量。由于SuperPCA涉及基因选择,其统计量的分布不再是标准的t分布,因此我们使用Gumbel极值分布的混合模型来估计p值。
- 实施流程
鉴于SuperPCA与AES-PCA在语法上的相似性,我们将重点介绍AES-PCA的实施步骤。需注意,当numReps参数设置为大于0的值时,AESPCA_pvals()函数会采用参数化方法来评估途径的统计显著性,具体模型为:“表型 ~ 截距 + 第一主成分(PC1)”。途径的p值通过似然比检验得出,该检验将所提模型与仅包含截距的零假设模型进行比较。
ovarian_aespcOut <- AESPCA_pVals(
# The Omics data container
object = ov_OmicsSurv,
# One principal component per pathway
numPCs = 1,
# Use parallel computing with 2 cores
parallel = TRUE,
numCores = 2,
# # Use serial computing
# parallel = FALSE,
# Estimate the p-values parametrically
numReps = 0,
# Control FDR via Benjamini-Hochberg
adjustment = “BH”
)

最终结果将包括每个途径的p值、基于AESPCA方法为每个样本估计的主成分得分,以及各蛋白质对这些主成分的贡献度。
- 途径分析结果
我们可以使用getPathpVals函数来获取AES-PCA在卵巢癌数据上识别出的前10条最重要的通路。
getPathpVals(ovarian_aespcOut, numPaths = 10)

在我们绘制p值图表之前,会先筛选出20个最重要的途径(默认设置中numPaths参数值为20)。
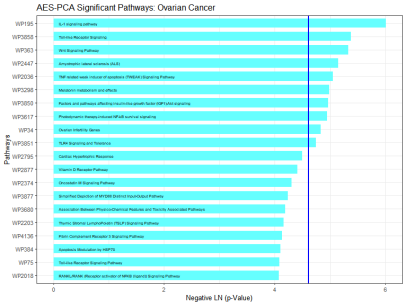
ovOutGather_df <- getPathpVals(ovarian_aespcOut, score = TRUE)
接下来,我们将为这些顶尖途径绘制它们的重要性评分图。图中的分数代表每个途径原始p值的负自然对数,以此来展示它们在统计学上的意义。
ggplot(ovOutGather_df) +
# set overall appearance of the plot
theme_bw() +
# Define the dependent and independent variables
aes(x = reorder(terms, score), y = score) +
# From the defined variables, create a vertical bar chart
geom_col(position = “dodge”, fill = “#66FFFF”, width = 0.7) +
# Add pathway labels
geom_text(
aes(
x = reorder(terms, score),
label = reorder(description, score),
y = 0.1
),
color = “black”,
size = 2,
hjust = 0
) +
# Set main and axis titles
ggtitle(“AES-PCA Significant Pathways: Ovarian Cancer”) +
xlab(“Pathways”) +
ylab(“Negative LN (p-Value)”) +
# Add a line showing the alpha = 0.01 level
geom_hline(yintercept = -log(0.01), size = 1, color = “blue”) +
# Flip the x and y axes
coord_flip()
- 提取相关基因
由于途径是预先设定的,每个途径中通常只有部分基因与特定表型相关,并影响途径的统计显著性。在AESPCA分析中,我们关注的是那些在第一主成分分析(AESPCs)中具有非零载荷的基因,因为它们与表型变化有直接联系。
以“IL-1信号途径”(Wikipathways WP195)为例,我们能够利用getPathPCLs()函数来提取相关的主成分及其对应的蛋白质载荷值。
wp195PCLs_ls <- getPathPCLs(ovarian_aespcOut, “WP195”)

非零载荷的蛋白质可以用以下方法提取:
wp195Loadings_df <- wp195PCLs_ls$Loadings %>% filter(PC1 != 0)

同时,我们可以将这些载荷数据整理,以便用于图形化展示。
wp195Loadings_df <-
wp195Loadings_df %>%
# Sort Loading from Strongest to Weakest
arrange(desc(abs(PC1))) %>%
# Order the Genes by Loading Strength
mutate(featureID = factor(featureID, levels = featureID)) %>%
# Add Directional Indicator (for Colour)
mutate(Direction = factor(ifelse(PC1 > 0, “Up”, “Down”)))
现在我们将使用ggplot2的geom_col()函数构造一个柱状图。
ggplot(data = wp195Loadings_df) +
# Set overall appearance
theme_bw() +
# Define the dependent and independent variables
aes(x = featureID, y = PC1, fill = Direction) +
# From the defined variables, create a vertical bar chart
geom_col(width = 0.5, fill = “#005030”, color = “#f47321”) +
# Set main and axis titles
labs(
title = “Gene Loadings on IL-1 Signaling Pathway”,
x = “Protein”,
y = “Loadings of PC1”
) +
# Remove the legend
guides(fill = FALSE)

- 个体化的主成分分析(PCA)
在探究复杂疾病时,由于疾病的成因及对治疗的反应在不同个体间存在显著差异,因此除了为所有患者确定与疾病相关的途径外,制定有效的(个性化的)治疗方案也需要了解特定途径在每个患者身上的异常情况。
为了实现这一目标,我们可以评估每个个体的途径活性。如同之前所述,getPathPCLs()函数不仅能够返回个体途径的主成分分析结果,还能提供个体特定的估计值。
ggplot(data = wp195PCLs_ls$PCs) +
# Set overall appearance
theme_classic() +
# Define the independent variable
aes(x = V1) +
# Add the histogram layer
geom_histogram(bins = 10, color = “#005030”, fill = “#f47321”) +
# Set main and axis titles
labs(
title = “Distribution of Sample-specific Estimate of Pathway Activities”,
subtitle = paste0(wp195PCLs_ls$pathway, “: “, wp195PCLs_ls$description),
x = “PC1 Value for Each Sample”,
y = “Count”)

这张图显示了不同患者之间在途径活性上可能存在相当大的差异。
- 提取重要途径的详细数据集
研究者们经常希望深入了解顶级途径分析背后的具体数据,尤其是那些与途径直接相关的特定基因。通过SubsetPathwayData()函数,我们可以提取这些数据。以下步骤展示了如何提取最具统计意义的途径(例如IL-1信号途径)的数据:
wp195Data_df <- SubsetPathwayData(ov_OmicsSurv, “WP195”)

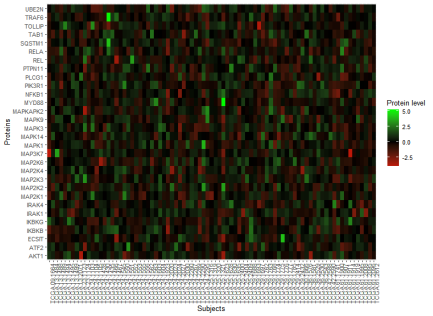
为了制作该途径内蛋白质的热图,我们首先需要将数据整理成三个主要列。
wp195gather_df <- wp195Data_df %>% arrange(EventTime) %>% select(-EventTime, -EventObs) %>% gather(protein, value, -sampleID)
现在,让我们来绘制热图:
ggplot(wp195gather_df, aes(x = protein, y = sampleID)) + geom_tile(aes(fill = value)) + scale_fill_gradient2(low = “red”, mid = “black”, high = “green”) + labs(x = “Proteins”, y = “Subjects”, fill = “Protein level”) +theme(axis.text.x = element_text(angle = 90)) + coord_flip()
我们还可以对途径内的单个基因进行深入分析:
library(survival)
NFKB1_df <- wp195Data_df %>% select(EventTime, EventObs, NFKB1)
wp195_mod <- coxph(Surv(EventTime, EventObs) ~ NFKB1,data = NFKB1_df)
summary(wp195_mod)

此外,我们能够为单个基因的高表达或低表达患者的群体估计Kaplan-Meier生存曲线:
# Add the direction
NFKB1_df <-NFKB1_df %>%
# Group subjects by gene expression
mutate(NFKB1_Expr = ifelse(NFKB1 > median(NFKB1), “High”, “Low”)) %>%
# Re-code time to years
mutate(EventTime = EventTime / 365.25) %>%
# Ignore any events past 10 years
filter(EventTime <= 10)
# Fit the survival model
NFKB1_fit <- survfit(Surv(EventTime, EventObs) ~ NFKB1_Expr, data = NFKB1_df)
最终,我们可以在NFKB1蛋白的表达水平上绘制这些生存曲线。
library(survminer)
ggsurvplot(NFKB1_fit,conf.int = FALSE, pval = TRUE,xlab = “Time in Years”,palette = c(“#f47321”, “#005030”),xlim = c(0, 10))
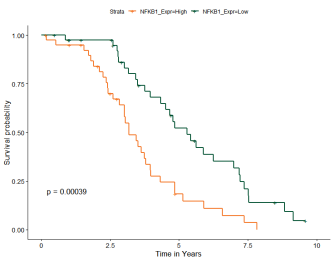
这张图展示了根据NFKB1蛋白表达水平(高或低)分组的患者的Kaplan-Meier生存曲线。从图中可以看出,高表达组(NFKB1_Expr=High)和低表达组(NFKB1_Expr=Low)的生存率有显著差异,这表明NFKB1可能是影响疾病预后的关键生物标志物。
伙伴们,小师妹已经带领大家一起完成了pathwayPCA的初步探索。现在,你可以开始自己动手,利用pathwayPCA包来分析基因表达数据啦,包括测试途径与表型之间的关联、提取相关基因、计算代表个体样本途径的主成分等。在使用过程中,记得充分利用pathwayPCA提供的图形函数,它可以让你的分析过程更加直观和高效!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~