嘿朋友们,欢迎来到生物信息学的精彩世界!面对RNA数据分析中的一个常见挑战——在rRNA和tRNA中识别修饰核苷酸的身份和位置,你是否有束手无策的时候?别担心,小师妹今天就带你探索RNAmodR.ML这个强大的R包,它运用前沿的机器学习技术,为我们揭示转录后修饰的复杂模式。
尽管转录后修饰广泛存在于rRNA和tRNA中,但要确定修饰核苷酸的具体身份和位置却非易事。传统方法,如逆转录(RT)过程中的引物延伸,虽然能够揭示某些修饰的端倪,但面对更深层次的分析就显得力不从心。幸运的是,随着高通量测序技术的发展,RNAmodR.ML应时而生,它通过训练机器学习模型来识别那些难以捉摸的修饰模式,为RNA修饰研究带来了新的可能。RNAmodR.ML是由Felix G.M. Ernst和Denis L.J. Lafontaine在2024年提出的机器学习模型的集大成者。它不仅能够帮助我们发现那些传统方法难以捉摸的修饰,还能预测未知的修饰位点,极大提升我们的研究效率。
请注意,RNAmodR.ML在处理大型数据集时可能需要较大的内存,因此小师妹建议使用服务器进行分析。如果您需要性价比高的服务器租赁服务,欢迎联系小师妹。
准备好了吗?接下来,小师妹将手把手带你从数据预处理到执行修饰检测,一步步领略RNAmodR.ML的魅力。如果你在分析的旅途中遇到任何难题,别担心,小师妹随时在这里为你解答疑惑。让我们携手揭开RNA修饰的神秘面纱,解码生命的奥秘吧!
首先,我们需要安装RNAmodR.ML包,可以通过以下命令进行安装:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“RNAmodR.ML”)
BiocManager::install(“RNAmodR.Data”)
安装完成后,我们可以通过以下命令加载RNAmodR.ML包:
library(rtracklayer)
library(GenomicRanges)
library(RNAmodR.ML)
library(RNAmodR.Data)
现在让我们来聊聊RNAmodR.ML包里的大明星——ModifierML类。这个类在RNAmodR包的基础上做了个升级,增加了一个叫mlModel的新空间,还有一对儿管理这个空间的助手:getMLModel和setMLModel。别忘了,还有个特殊的函数useMLModel,它能在我们需要的时候被召唤出来。
现在,不管你用的是哪种机器学习模型,ModifierMLModel都能搞定。比如,如果你喜欢用ranger包(Wright和Ziegler,2017),那就有ModifierMLranger;如果你偏好keras包(Allaire和Chollet,2018),那就有ModifierMLkeras。
接下来,我会带你一步步体验如何用RNAmodR.ML包来打造你自己的机器学习模型。从准备数据到训练模型,每个步骤我都会详细讲解,保证让你对RNAmodR.ML包的强悍功能了如指掌。别担心,跟着小师妹,我们一起乘风破浪!
一、创建新的Modifier类
在这个教程中,我们要来点实际的挑战,比如在AlkAnilineSeq数据中检测D位置。首先,我们要定义一个特定的ModifierML类来加载pileup和coverage数据。在这里,我们会用到专门针对RNA的RNAModifierML类。
setClass(“ModMLExample”,
contains = c(“RNAModifierML”),
prototype = list(mod = c(“D”),
score = “score”,
dataType = c(“PileupSequenceData”,
“CoverageSequenceData”),
mlModel = character(0)))
# constructor function for ModMLExample
ModMLExample <- function(x, annotation = NA, sequences = NA, seqinfo = NA,
…){
RNAmodR:::Modifier(“ModMLExample”, x = x, annotation = annotation,
sequences = sequences, seqinfo = seqinfo, …)
}
setClass(“ModSetMLExample”,
contains = “ModifierSet”,
prototype = list(elementType = “ModMLExample”))
# constructor function for ModSetMLExample
ModSetMLExample <- function(x, annotation = NA, sequences = NA, seqinfo = NA,
…){
RNAmodR:::ModifierSet(“ModMLExample”, x, annotation = annotation,
sequences = sequences, seqinfo = seqinfo, …)
}
现在,因为mlModel这个位置现在是空的,所以创建这个对象的时候,并不会自动开始寻找修饰。但是,它会按照我们想要的格式来整理数据。记住,aggregate_example函数只是个示例,数据的聚合其实是模型构建的一部分。
setMethod(
f = “aggregateData”,
signature = signature(x = “ModMLExample”),
definition =
function(x){
aggregate_example(x)
}
)
- 获取训练数据
我们可以通过下面的代码来下载需要的数据:
proxy <- httr::use_proxy(Sys.getenv(‘http_proxy’))
httr::set_config(proxy)
ExperimentHub::setExperimentHubOption(“PROXY”, proxy)
annotation <- GFF3File(RNAmodR.Data.example.gff3())
sequences <- RNAmodR.Data.example.fasta()
files <- list(“wt” = c(treated = RNAmodR.Data.example.bam.1(),
treated = RNAmodR.Data.example.bam.2(),
treated = RNAmodR.Data.example.bam.3()))
接着,我们只需要创建一个ModMLExample对象,然后让它开始工作。
me <- ModMLExample(files[[1]], annotation, sequences)
接下来,我们需要加载或创建一些已知被修饰的位置坐标,同时,我们也需要那些已知没有被修饰的位置坐标。
data(“dmod”,package = “RNAmodR.ML”)
# we just select the next U position from known positions
nextUPos <- function(gr){
nextU <- lapply(seq.int(1L,2L),
function(i){
subseq <- subseq(sequences(me)[dmod$Parent], start(dmod)+3L)
pos <- start(dmod) + 2L +
vapply(strsplit(as.character(subseq),””),
function(y){which(y == “U”)[i]},integer(1))
ans <- dmod
ranges(ans) <- IRanges(start = pos, width = 1L)
ans
})
nextU <- do.call(c,nextU)
nextU$mod <- NULL
unique(nextU)
}
nondmod <- nextUPos(dmod)
nondmod <- nondmod[!(nondmod %in% dmod)]
coord <- unique(c(dmod,nondmod))
coord <- coord[order(as.integer(coord$Parent))]
有了这些坐标,我们就可以使用trainingData函数,把ModMLExample对象聚合的数据提取出一个训练数据集。
trainingData <- trainingData(me,coord)
trainingData <- unlist(trainingData, use.names = FALSE)
# converting logical labels to integer
trainingData$labels <- as.integer(trainingData$labels)
简单来说,这个过程就像是在准备烹饪的食材。我们先定义好我们的“食材”——也就是那些数据坐标,然后用这些“食材”来制作我们的“大餐”——训练数据集。
- 模型的训练
跟着小师妹,我们直接来看最精彩的部分——训练模型!仅需一行代码,我们就可以实现用ranger包提供的功能来训练一个随机森林模型。
library(ranger)
model <- ranger(labels ~ ., data.frame(trainingData))
四、创建一个新的ModifierMLModel
现在,我们已经训练好了模型,接下来就是用它来创建一个新的ModifierMLModel类和对象。
setClass(“ModifierMLexample”,
contains = c(“ModifierMLranger”),
prototype = list(model = model))
ModifierMLexample <- function(…){
new(“ModifierMLexample”)
}
mlmodel <- ModifierMLexample()
为了让这个模型能在ModifierMLModel类中发挥作用,我们还需要定义一个访问器来获取模型做出的预测。这个访问器叫做useModel,而且在上面提到的“使用RNAmodR.ML”那部分,已经为我们预定义好了。
getMethod(“useModel”, c(“ModifierMLranger”,”ModifierML”))
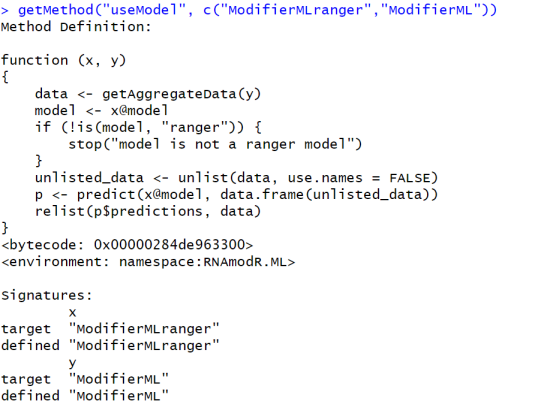
如果这个函数的结果对某个特定的模型不适用,没关系,你可以根据自己的需要来重新定义它。RNAmodR.ML定义的这个函数会返回一个NumericList,这个列表会和ModifierML对象的聚合数据一起提供。
五、模型的使用
现在,我们已经生成了ModifierMLexample这个包装器,接下来我们可以用setMLModel函数把它设置到ModifierML对象上。不过,如果模型已经被保存为包的一部分,那么这个步骤就不是必须的了,因为它可以作为类定义的一部分。
setMLModel(me) <- mlmodel
为了让预测数据能够被使用,我们需要实现另一个函数来保存预测结果到聚合数据中。这个函数就叫做useMLModel。
setMethod(f = “useMLModel”,
signature = signature(x = “ModMLExample”),
definition =
function(x){
predictions <- useModel(getMLModel(x), x)
data <- getAggregateData(x)
unlisted_data <- unlist(data, use.names = FALSE)
unlisted_data$score <- unlist(predictions)
x@aggregate <- relist(unlisted_data,data)
x
}
)
通过重新运行聚合函数并强制更新数据,预测就会被执行并用来填充分数列,就像上面提到的那样。
me <- aggregate(me, force = TRUE)
六、模型的评估
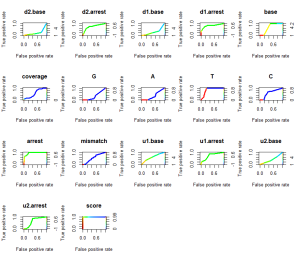
在构建模型的过程中,我们通常会包含某种形式的性能评估。除了这些特定于模型的测量方法,RNAmodR.ML还从RNAmodR包中继承了ROCR包的功能。这样,我们就可以在整个训练集或任何坐标上评估模型的性能了。
plotROC(me, dmod)
- ModifierML类的使用
因为我们打算用ModifierML对象来检测修饰,我们需要定义一个findMod函数。根据性能信息,我们设定了预测分数为0.8的阈值,用来检测D修饰。在下面的例子里,这个阈值被硬编码进了find_mod_example函数,但也可以用设置函数来实现。
setMethod( f = “findMod”,signature = signature(x = “ModMLExample”),definition =function(x){find_mod_example(x, 25L)})
现在,我们可以重新定义ModMLExample类,并且mlModel槽位已经设置为ModifierMLexample类。这样一来,ModMLExample类就大功告成了。
rm(me)
setClass(“ModMLExample”,contains = c(“RNAModifierML”),prototype = list(mod = c(“D”),score = “score”,dataType = c(“PileupSequenceData”, “CoverageSequenceData”), mlModel = “ModifierMLexample”))
me <- ModMLExample(files[[1]], annotation, sequences)
检测到的修饰可以通过modifications函数来访问。
mod <- modifications(me)
mod <- split(mod, factor(mod$Parent,levels = unique(mod$Parent)))
mod


八、模型的优化
有些检测到的修饰看起来是靠谱的,但也有一些位置似乎是噪声。
options(ucscChromosomeNames=FALSE)
plotDataByCoord(sequenceData(me),mod[[“4”]][1])
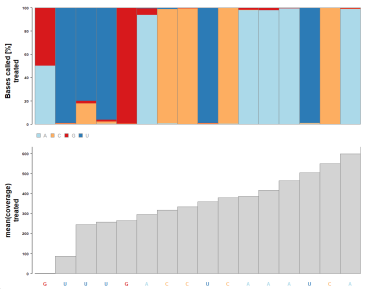
别担心,我们有几种方法可以优化模型:我们可以提高预测分数的阈值,比如提高到0.9,或者可以把这些位置作为未修饰的位置加入到另一个训练数据集中重新训练模型。此外,通过提高测序深度,我们通常也能改善训练数据的质量。
nonValidMod <- mod[c(“1″,”4”)]
nonValidMod[[“18”]] <- nonValidMod[[“18”]][2]
nonValidMod[[“26”]] <- nonValidMod[[“26”]][2]
nonValidMod <- unlist(nonValidMod)
nonValidMod <- nonValidMod[,”Parent”]
coord <- unique(c(dmod,nondmod,nonValidMod))
coord <- coord[order(as.integer(coord$Parent))]
比如说,我们可以训练一个新的模型,把之前模型错误识别的位置作为未修饰的位置包含进来。
trainingData <- trainingData(me,coord)
trainingData <- unlist(trainingData, use.names = FALSE)
trainingData$labels <- as.integer(trainingData$labels)
model2 <- ranger(labels ~ ., data.frame(trainingData), num.trees = 2000)
setClass(“ModifierMLexample2”,contains = c(“ModifierMLranger”),prototype = list(model = model2))
ModifierMLexample2 <- function(…){new(“ModifierMLexample2”)}
mlmodel2 <- ModifierMLexample2()
me2 <- me
setMLModel(me2) <- mlmodel2
me2 <- aggregate(me2, force = TRUE)
更新了ModifierMLexample类并重新聚合数据后,模型的性能看起来有所提高,并且在检测D修饰方面得到了更好的结果。
plotROC(me2, dmod, score=”score”)
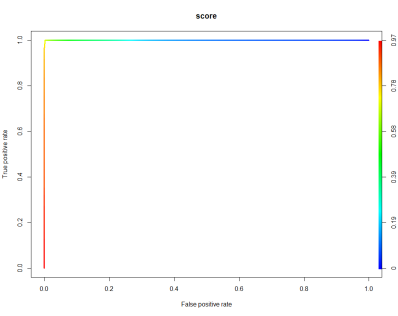
有些位置不再被检测到,这表明当前模型还不是最优解,正如上文建议的,我们还可以进一步改进其他因素。
setMethod(f = “findMod”,signature = signature(x = “ModMLExample”),definition =function(x){find_mod_example(x, 25L)})
me2 <- modify(me2, force = TRUE)
modifications(me2)
除了训练单个模型,我们还可以训练多个模型并将它们组合成一个ModifierSet。
mse <- ModSetMLExample(list(one = me, two = me2))
然后,我们可以分析多个模型的整体性能,或者比较它们的个体性能。
plotROC(mse, dmod, score= “score”,
plot.args = list(avg = “threshold”, spread.estimate = “stderror”))

如果训练了多个模型,而且每个模型都提供了有用的信息,那么这些模型可以被打包进一个ModifierMLModel类中,以组合多个模型的输出。比如,上面提到的useMLModel和/或useModel函数可能需要被修改,以提供检测修饰的一个或多个分数。
伙伴们,跟着小师妹的引导,我们已经顺利完成了RNAmodR.ML包的探索之旅。现在,是时候轮到你们大显身手了。动手使用RNAmodR.ML包来分析那些复杂的基因表达数据吧,无论是检测RNA分子上的修饰位点,还是构建自己的机器学习模型,RNAmodR.ML都能助你一臂之力。
记得在使用过程中,充分利用RNAmodR.ML提供的各项功能,比如自定义ModifierML类,或者使用不同的机器学习模型进行预测。同时,别忘了运行性能评估,这会让你对模型的准确性和可靠性有更直观的了解。
如果你在分析的道路上遇到任何障碍,或者想要进一步优化模型,随时回来找小师妹。我们一起讨论,一起进步。现在,就让我们带着新学的技能,勇敢地迈向生物信息学的新高峰吧!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~