
大家好!今天大海哥要向大家介绍一个在基因表达数据分析领域中非常重要的R包——Mfuzz包!Mfuzz包是专门用于分析基因表达数据的强大工具。要知道,基因表达调控的复杂性和连续性使得传统的硬聚类方法无法完全揭示其中的规律,而Mfuzz采用的模糊C均值聚类算法(Fuzzy c-means clustering)则能够有效地进行软聚类分析,提供了更为细腻和准确的基因表达模式解析。大海哥可是厉害的生信高手,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!
在本次学习中,我们不仅将学会如何安装和使用Mfuzz包,还将掌握基因表达数据的预处理、标准化、以及软聚类分析的方法。通过实际示例的演示,我们将更深入地理解基因表达的动态变化和复杂调控机制,为我们解读基因功能提供重要线索。我相信,通过今天的学习,同学们都能够更加熟练地应用Mfuzz包,为基因表达数据分析的研究进步贡献自己的一份力量!接下来,同学们跟紧大海哥的步伐,让我们正式开启对Mfuzz包的学习之旅吧!
本次介绍的工具需要在服务器上才能正常运行,同学们如果没有自己的服务器欢迎联系我们进行服务器租赁~
Mfuzz包介绍
Mfuzz是R语言中的一个专门用于基因表达数据分析的包,它采用模糊C均值聚类算法,对基因表达数据进行软聚类。与传统的硬聚类方法不同,软聚类允许每个基因分配到多个聚类中,每个基因对不同聚类的隶属度值介于0到1之间。这种方法更好地反映了基因表达调控的复杂性和连续性。通过Mfuzz,用户可以生成详细的聚类图,展示基因在不同时间点或条件下的表达模式。这些功能使得Mfuzz在基因表达研究中具有广泛应用,帮助研究人员揭示基因调控机制和生物学过程的动态变化。
Mfuzz包的安装
需要R语言版本为4.4,在控制台中输入以下命令:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager “)
BiocManager::install(“Mfuzz”) # 在BiocManager环境下安装Mfuzz
查看是否安装成功
packageVersion(“Mfuzz”) # 查看Mfuzz版本

显示为2.64.0版本,则表示已经安装了Mfuzz包。
数据预处理
载入需要的包和数据
首先,我们需要载入本次学习的主角——Mfuzz包。
library(Mfuzz) #载入Mfuzz包
在本文中,大海哥将使用酵母细胞中3000个基因17个时间点的表达强度作为示例数据。我们需要载入数据并进行一系列预处理,代码如下:
data(yeast) #载入示例数据yeast
yeast #查看数据信息
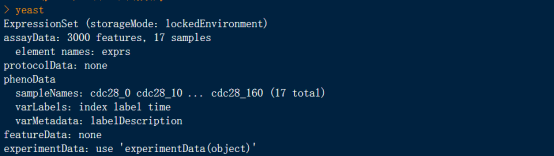
显示如上图,可知共3000个基因,17个时间点。
剔除缺失值以及过滤表达水平低或仅表现出小变化的基因
第一步,我们排除测量值缺失超过 25% 的基因。需要注意,基因表达矩阵中缺失值应标记为 NA。
yeast.r <- filter.NA(yeast, thres=0.25)
# 过滤掉缺失值(NA值)比例超过25%的基因数据
模糊 c 均值(Fuzzy c-means)与许多其他聚类算法一样,不允许缺失值。因此,我们将剩余的缺失值替换为相应基因的平均表达值。
yeast.f <- fill.NA(yeast.r, mode=”mean”)
# 填补缺失值(NA值),使用均值填补
大多数聚类分析都包括一个过滤步骤,以去除表达水平低或仅表现出小变化的基因。
tmp <- filter.std(yeast.f, min.std=0)
# 过滤标准差为0的基因数据,去除变化不大的基因
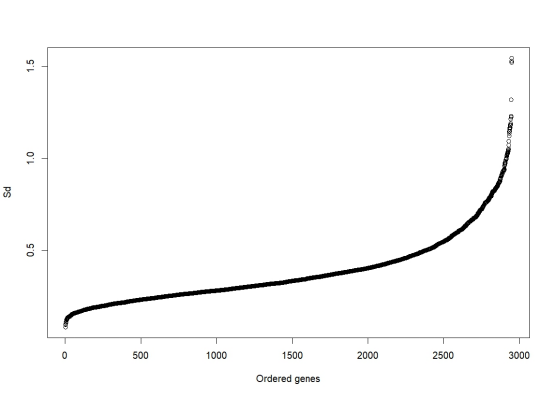
结果如上图所示,可知绝大多数基因的标准差均小于1,基因在不同时间节点的表达强度差别不大。
数据标准化
由于聚类是在欧几里得空间中进行的,因此基因的表达值标准化为均值为零,标准差为一。这一步确保了表达变化相似的基因向量在欧几里得空间中是接近的:
yeast.s <- standardise(yeast.f)
# 对基因表达数据进行标准化处理
重要的是,Mfuzz 假定给定的表达数据已经完全预处理,包括任何数据归一化。需要注意函数 standardise 不能替代归一化步骤,归一化是为了使不同样本具有可比性,而标准化是为了使转录物(基因或蛋白质)具有可比性。
基因表达数据的软聚类分析
聚类常用于揭示基因表达背后的调控机制。基因的调控通常不是“开-关”的模式,而是逐渐变化的方式,这允许对基因功能进行更精细的控制。聚类算法应当反映这一发现,区分一个基因在多大程度上跟随主要聚类模式。软聚类在这一任务中表现良好,因为它可以为基因i分配一个逐渐变化的成员值 µij 到聚类 j。成员值可以在0和1之间连续变化,使软聚类能够提供关于基因表达数据结构的更多信息。
对预处理之后的数据进行软聚类的代码如下:
cl <- mfuzz(yeast.s, c=16, m=1.25)
# 使用mfuzz进行模糊c均值聚类,并设置聚类数为16,模糊指数m为1.25
mfuzz.plot(yeast.s, cl=cl, mfrow=c(4,4), time.labels=seq(0,160,10))
# 使用mfuzz.plot绘制聚类结果,参数说明:yeast.s – 基因表达数据集cl – 聚类结果对象,mfrow=c(4,4) – 图形布局为4行4列,time.labels=seq(0,160,10) – 设置时间标签,从0到160,间隔为10
结果如下图所示:

由上图可知,每个子图代表一个基因聚类,横轴为时间(以分钟为单位),纵轴为基因表达变化。共有16个聚类,每个聚类包含若干个在相似时间模式下表达的基因。这些聚类展示了基因表达水平在不同时间点的动态变化。通过模糊C均值聚类算法(Fuzzy c-means clustering),基因被分配到多个聚类中,每个基因对于不同的聚类有不同的隶属度(membership value),范围从0到1。这种方法允许一个基因属于多个聚类,反映了基因表达调控的复杂性和连续性,而不是简单的二分法。
以上就是对于Mfuzz包的全部介绍了。通过本文,我们了解了Mfuzz作为高效的基因表达数据分析工具的强大功能和应用场景。Mfuzz采用模糊C均值聚类算法,能够有效地进行软聚类分析,揭示基因调控的复杂性和连续性。希望同学们能够继续深入学习和探索Mfuzz,通过不断实践和应用,大家一定能在基因表达数据分析的研究中取得更大的进步和成果。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!