前言
今天小果又和大家见面了,跟随小果的步伐来给自己空瘪的皮囊注入点知识的能量吧!接触单细胞分析的小伙伴们都知道亚簇分析是改善单细胞RNA-Seq数据聚类和表征的有力手段。然而,目前还没有工具可以系统地整合来自多个子集群的结果,这为下游分析中的准确数据量化、可视化和解释带来了障碍。所以今天小果给你介绍的R包Ragas一定要睁大眼睛好好学一下哦!它集成了多级子聚类对象,用于简化分析和可视化。实施了一种新的数据结构,以无缝连接和组装来自不同子聚类级别的各种单细胞分析,以及几个新的或增强的可视化功能。此外,还开发了一种重新投影算法来整合来自多个亚簇的最近邻图,以最大限度地提高它们在组合细胞嵌入上的可分离性,从而显着改善了稀有和均质亚群的呈现。此外,Ragas包有与Seurat对象兼容的统一数据结构,以协调单个单元数据的分析和可视化,是不是用起来更方便了呢?练了十年生信分析的小果对于单细胞的知识已经如鱼得水,相信有小果的加持一定能让大家学会!没有时间学习且有单细胞分析的小伙伴们快来滴滴小果噢!从分析到可视化直到你满意为止!
单细胞的数据实在是太大了,一个样本要吃掉你2-4G的内存,电脑吃不消的小伙伴们建议使用服务器,欢迎大家联系小果,保证服务器的性价比最高哦!
代码实现
加载R包并构建Pi对象
##本实验所使用的数据集已经集成在R包Ragas里面了,小伙伴们直接加载R包读取数据就可以啦。
##首先,加载 Ragas 包
library(Ragas)
csle.pbmc.small
## An object of class Seurat
## 1503 features across 3000 samples within 1 assay
## Active assay: RNA (1503 features, 1501 variable features)
## 3 dimensional reductions calculated: pca, harmony, umap
上面的 Seurat 对象是 Ragas 中的示例对象之一 包,这是 scRNA-Seq 数据分析的一部分,PBMC 来自 健康 (cHD) 和系统性红斑狼疮 (cSLE) 儿童 (https://www.nature.com/articles/s41590-020-0743-0)。执行 Ragas 分析,我们需要创建一个基于Seurat对象。集成后对象是单细胞数据对象已规范化、批量集成并执行 UMAP 和 聚类分析。注意:在我们的分析中,我们使用 Harmony 进行批量集成。
出于演示目的,我们预先构建了一个名为 Pi 的对象 “csle.pbmc.pi”基于示例 Seurat 对象 (csle.pbmc.small), 它与 Ragas 包一起发布,作为示例数据的一部分。
csle.pbmc.pi
## An object of class Pi
## 6 fields in the object: seurat.obj, exp.freq, markers, ds, cell.prop, parent.meta.data.
## The following fields have been processed:
## seurat.obj: A Seurat object of 1503 features and 2901 cells.
## 1 assay: RNA, and 4 reductions: pca, harmony, umap, rp
## exp.freq: A list of numeric matrices containing per gene expression frequencies
## 1 analysis run:
## ExpFreq|subcluster_idents|cutoff=0
## markers: A list of data frames containing marker results.
## 1 analysis run:
## Markers|subcluster_idents|AllMarkers|test.use=wilcox
## ds: A list of lists and data frames containing pseudobulk analysis results.
## 1 analysis run:
## DS|subcluster_idents|edgeR|group=Groups;sample=Names;gp1=cSLE;gp2=cHD;contrast=cSLE-cHD
## cell.prop: A list of data frames containing cell proportion analysis results
## 1 analysis run:
## CellProp|subcluster_idents|unpooled|unpool_by_Names|group=Groups
## Metadata from the parent object provided? No
## Subclusters integrated? Yes
my.pbmc.pi <- CreatePostIntegrationObject(object = csle.pbmc.small)
## Post-integration object created
my.pbmc.pi
## An object of class Pi
## 6 fields in the object: seurat.obj, exp.freq, markers, ds, cell.prop, parent.meta.data.
## The following field has been processed:
## seurat.obj: A Seurat object of 1503 features and 3000 cells.
## 1 assay: RNA, and 3 reductions: pca, harmony, umap
## Metadata from the parent object provided? No
## Subclusters integrated? No
上面的代码创建了一个仅包含 Seurat 的“最小”Pi 对象 对象。如果用户具有子集群中的其他对象 分析,可以遵循子聚类分析 用于将优化的子集群标识重新投影到 PBMC 的 Vignette。创建 Pi 对象后,可以先运行 UMAP 并检查 对象中所有单元格的簇。
RunDimPlot(object = my.pbmc.pi)
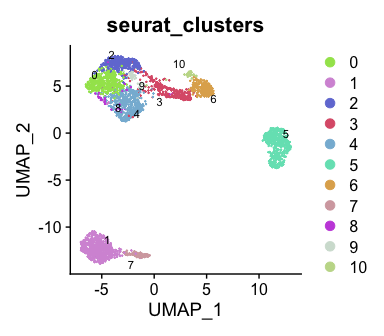
RunDimPlot(object = my.pbmc.pi,
group.by = “cluster.annotation”)

分析数据
Ragas 提供了一个用户友好的界面来集成和可视化 多种单细胞分析功能,包括 Marker 分析,使用伪散体法进行差分状态分析,以及差分细胞比例分析。
标记分析和矩阵图:用户可以通过运行 包装函数 RunFindAllMarkers,用于存储标记结果 在 Pi 对象中,以便以后可视化。
my.pbmc.pi <- RunFindAllMarkers(my.pbmc.pi,
ident = “cluster.annotation”)
my.pbmc.pi
运行 RunFindAllMarkers 后,一个名为“markers”的新数据字段将 被添加到当前 Pi 对象中,该对象包含具有长 唯一标识符称为 “标记“cluster.annotation|AllMarkers|test.use=wilcox“。这个独一无二的 标识符可用于检索标记结果。
my.pbmc.pi[[“markers”]]
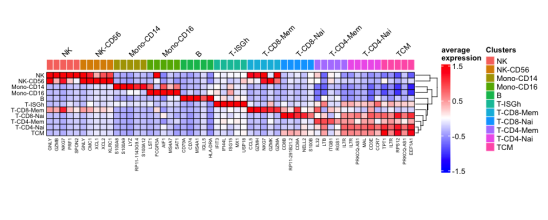
矩阵图:标记分析完成后,每个聚类的顶级标记可以使用矩阵图绘制
RunMatrixPlot(my.pbmc.pi,
markers.key = “Markers|cluster.annotation|AllMarkers|test.use=wilcox”,
column.anno.name.rot = 45,
heatmap.height = 6)
伪体积分析
单细胞研究的另一项重要任务是识别 各簇间差异表达基因,也称为差分状态 (DS) 分析。我们使用 函数从 Muscat 包到对多样本多组scRNA-Seq进行伪批量分析设计。
在运行伪批量分析之前,我们需要先进行表达 频率分析。对给定簇的低表达基因 将在最终的伪批量输出中被过滤掉。
my.pbmc.pi <- CalculateExpFreqs(my.pbmc.pi,
ident = “cluster.annotation”,
verbose = FALSE)
my.pbmc.pi
#批量分析
my.pbmc.pi <- RunPseudobulkAnalysis(object = my.pbmc.pi,
ident.var = “cluster.annotation”,
group.var = “Groups”,
sample.var = “Names”,
group.1 = “cSLE”,
group.2 = “cHD”)
my.pbmc.pi
#使用 DSDotPlot 可视化伪批量结果
RunDSDotPlot(object = my.pbmc.pi,
exp.freq.key = “ExpFreq|cluster.annotation|cutoff=0”,
ds.key = “DS|cluster.annotation|edgeR|group=Groups;sample=Names;gp1=cSLE;gp2=cHD;contrast=cSLE-cHD”,
p.filter = 0.05,
FC.filter = 4,
to.adjust = TRUE
)

比例分析
Ragas还提供了一组丰富的函数来可视化单元格比例数据。比例分析有两种方法:池化和非池化。混合比例分析在单细胞研究中得到了广泛的应用,但它的主要缺点是由于混合了所有样本的所有细胞而失去了对生物复制的跟踪,这使得很难评估细胞比例的可变性。因此,对于多样本多组设计,非池化是首选。
#使用“池化”图时,用户可以进一步选择其中之一 以下两种方法计算比例:按“聚类”或按 “组”。
RunProportionPlot(csle.pbmc.small, ## input can be a Seurat object
ident = “cluster.annotation”,
group.by = “Groups”,
method = “pooled”,
pooled.prop.by = “cluster”, ## default
axis.text.angle = 45,
axis.text.size = 8,
return.value = ‘ggplot’
)
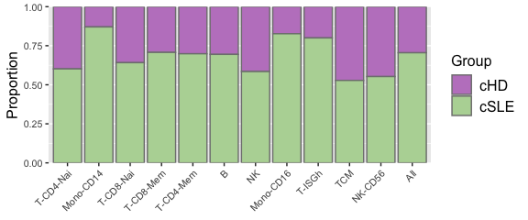
如果我们交换参数“ident”和“group.by”的输入,这将 导致显示所有聚类的单元格比例的不同条形图 由每组中的细胞总数归一化。这是 本质上是“按组”合并单元格比例的堆叠版本,但条形图是堆叠的。
my.pbmc.pi <- RunProportionPlot(my.pbmc.pi, ## if input is a Pi object, an updated Pi object will be the returned by default
ident = “Groups”,
group.by = “cluster.annotation”,
method = “pooled”,
pooled.prop.by = “cluster”, ## default
axis.text.size = 8)
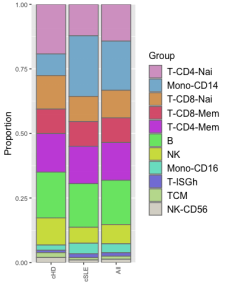
在另一种情况下,用户可以按组绘制单元格比例, 这意味着给定集群中给定组的细胞计数为由每组的细胞总数归一化。
my.pbmc.pi <- RunProportionPlot(my.pbmc.pi,
ident = “cluster.annotation”,
group.by = “Groups”,
method = “pooled”,
pooled.prop.by = “group”,
axis.text.size = 8)

未池化比例图
未合并比例图适用于多样本、多组 研究设计,允许比较感兴趣的组,以及评估它们在内部的变异性每个组。
my.pbmc.pi <- RunProportionPlot(my.pbmc.pi,
ident = “cluster.annotation”,
group.by = “Groups”,
method = “unpooled”,
unpool.by = “Names”,
unpool.ncol = 3,
title.text.size = 6)

#未池化条形图
my.pbmc.pi <- RunProportionPlot(my.pbmc.pi,
ident = “cluster.annotation”,
group.by = “Groups”,
method = “unpooled”,
unpool.by = “Names”,
unpool.plot.type = “barplot”,
unpool.ncol = 3,
title.text.size = 6)

其他可视化
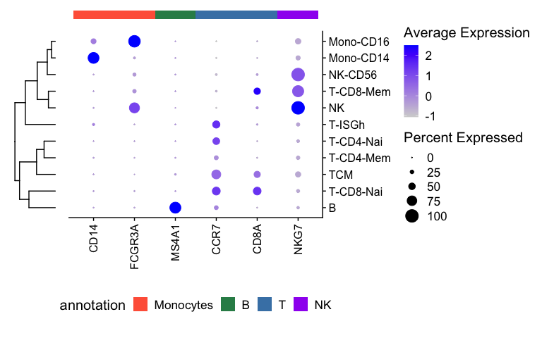
#带注释的点阵图
my.list <- list(Monocytes = c(“CD14″,”FCGR3A”),
B = “MS4A1”,
T = c(“CCR7”, “CD8A”),
NK = “NKG7”)
RunAnnotatedDotPlot(object = my.pbmc.pi,
annotations = my.list,
annotation.cols = c( ‘Monocytes’= ‘tomato’, ‘B’ = ‘seagreen’, ‘T’ = ‘steelblue’, ‘NK’ = ‘purple’),
group.by = “cluster.annotation”)
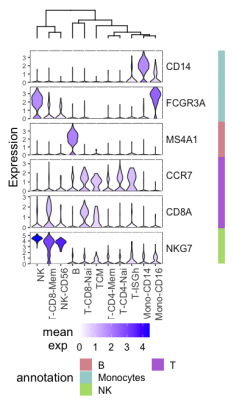
#堆叠小提琴图
RunStackedVlnPlot(my.pbmc.pi,
ident = “cluster.annotation”,
features = my.list,
color.by = “mean.exp”,
column.names.rotation = 90)
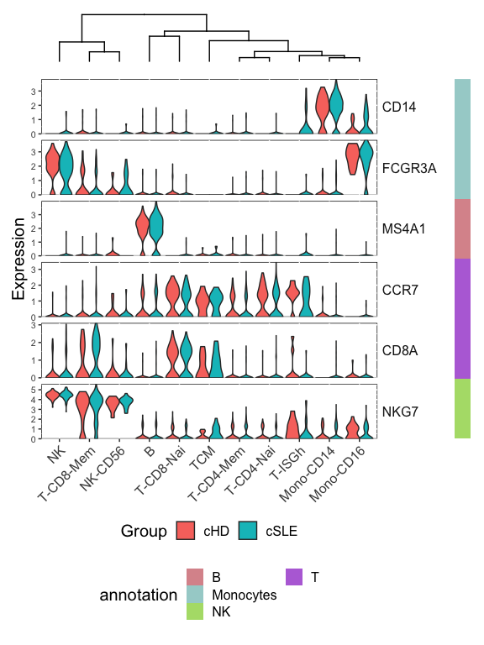
RunStackedVlnPlot(my.pbmc.pi,
ident = “cluster.annotation”,
features = my.list,
split.by = “Groups”,
color.by = “split.var”,
column.names.rotation = 45)
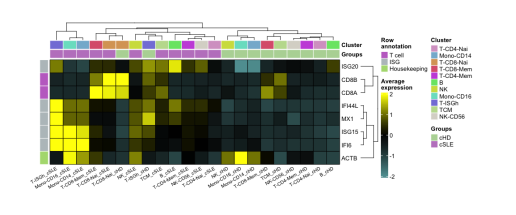
##汇总热图
features <- list(“T cell” = c(“CD8A”, “CD8B”),
ISG = c(“ISG15″,”ISG20″,”IFI44L”, “IFI6”, “MX1”),
“Housekeeping” = c(“ACTB”))
RunSummarizedHeatmap(object = my.pbmc.pi,
ident = “cluster.annotation”,
features = features,
split.by = “Groups”,
column.names.rotation = 30,
heatmap.width = 20)
##表达式图
RunExpressionPlot(object = my.pbmc.pi,
feature = “ISG15”,
ident = “cluster.annotation”,
group.by = “Names”,
split.by = “Groups”)

数据导出
在 Ragas 中,我们提供了 ExportPiData 函数来导出分析结果。
ExportPiData(object = csle.pbmc.pi,
field = ‘ds’,
key = ‘DS|subcluster_idents|edgeR|group=Groups;sample=Names;gp1=cSLE;gp2=cHD;contrast=cSLE-cHD’,
file.prefix = ‘pbmc.ds’ ## A mandatory argument. User should provide an informative prefix (e.g., “csle.pbmc”) for exported data
)
小结
小果向大家介绍了Ragas,这是一个基于R的框架,用于单细胞子集群数据的集成分析和可视化。Ragas提供了三个主要贡献:(i)定义了一个与Seurat对象兼容的统一数据结构,以协调单个单元数据的分析和可视化。(ii)开发了一种重新投影算法,将来自多个亚簇的细胞包埋组合在一起,该算法可以显示足够的结构粒度,用于稀有或同质亚群。(iii) 作为对现有Seurat可视化功能的补充,Ragas 提供了一个直观的界面来生成新的或增强的单细胞图。到这里小果要和大家说再见了!最后小果给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!