前言
小伙伴们都趋向于使用未校正批次效应后的数据进行差异基因分析,但是在富集分析的过程中,使用校正后的数据还是未校正批次效应后的数据呢?校正批次效应后的数据会不会掩盖部分真实的生物学差异?样本之间的批次效应是否会影响基因富集分析结果仍然是一个争论。今天大海哥给大家介绍的方法重新审视现有的、主流的基因集富集方法,找到受批次效应影响较小的基因集富集分析方法。像小伙伴们熟悉的AUCell、UCell、singscore、ssGSEA、JASMINE 和 Viper被打包到R包irGSEA。通过采用鲁棒秩聚合算法,irGSEA合并了所有六种方法的结果,以确定目标基因集在不同评分方法中的统计学意义。irGSEA采用基于批量的评分方法可以减少仅采用单细胞评分方法引入的偏差。该框架结合了RRA方法,以在各种评分方法中识别显着富集的基因集。
irGSEA要点:
- irGSEA通过采用鲁棒的秩聚合算法来合并所有六种基于秩的方法的结果,以确定目标基因集在不同评分方法中的统计学意义。
- irGSEA支持19种基因集评分方法,并集成了多种用户友好的可视化工具来显示结果。
- irGSEA有助于评估细胞簇内可能具有重要意义的生物过程,强调其在探索细胞功能方面的实用性,而不是在细胞类型鉴定方面表现出色。
大海哥就是这么的宠粉,有什么生信分析上的问题大家尽管咨询大海哥!没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找大海哥哦!练了十年生信分析的大海哥对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!

生信数据处理起来占用内存实在太大了,放过自己的电脑吧!大海哥在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系大海哥,保证服务器的性价比最高哦!
代码教程
1.安装前的准备
R包irGSEA淘汰了所有需要考虑样本组成的基因集富集分析方法,选取了基于单个样本的基因表达排名的基因集分析方法:AUCell、UCell和singscore。同时,也部分改进了ssGSEA,取消最后的标准化步骤,使之更贴近于单个样本的基因集富集分析。
在安装irGSEA 需要查看自己是否安装了以下这些包。
# 从 CRAN安装R包
cran.packages <- c(“msigdbr”, “dplyr”, “purrr”, “stringr”,”magrittr”,
“RobustRankAggreg”, “tibble”, “reshape2”, “ggsci”,
“tidyr”, “aplot”, “ggfun”, “ggplotify”, “ggridges”,
“gghalves”, “Seurat”, “SeuratObject”, “methods”,
“devtools”, “BiocManager”,”data.table”,”doParallel”,
“doRNG”)
if (!requireNamespace(cran.packages, quietly = TRUE)) {
install.packages(cran.packages, ask = F, update = F)
}
#从Bioconductor安装R包
bioconductor.packages <- c(“GSEABase”, “AUCell”, “SummarizedExperiment”,
“singscore”, “GSVA”, “ComplexHeatmap”, “ggtree”,
“Nebulosa”)
if (!requireNamespace(bioconductor.packages, quietly = TRUE)) {
BiocManager::install(bioconductor.packages, ask = F, update = F)
}
#从Github安装R包
if (!requireNamespace(“UCell”, quietly = TRUE)) {
devtools::install_github(“carmonalab/UCell”)
}
if (!requireNamespace(“irGSEA”, quietly = TRUE)) {
devtools::install_github(“chuiqin/irGSEA”)
}
2.加载示例数据
我们允许直接输入单细胞表达矩阵或者Seurat对象。我们内置了Seurat包,可以将多种基因集的富集分数矩阵直接保存到Seurat对象中。同时,我们也支持过滤单细胞表达矩阵中所有细胞表达量为0的基因。当然,用户也可以自定义自己的过滤标准。合适的过滤指标可以改善富集分析的结果。
我们通过SeuratData包加载示例数据集(注释好的PBMC数据集)
# devtools::install_github(‘satijalab/seurat-data’)
library(SeuratData)
# view all available datasets
View(AvailableData())
# download 3k PBMCs from 10X Genomics
# 示例数据有点大,建议在网速好的时候下载,如果失败了,请重启R后多尝试几遍
InstallData(“pbmc3k”)
# the details of pbmc3k.final
?pbmc3k.final
library(Seurat)
library(SeuratData)
#加载数据
data(“pbmc3k.final”)
pbmc3k.final <- UpdateSeuratObject(pbmc3k.final)
#绘图
DimPlot(pbmc3k.final, reduction = “umap”,
group.by = “seurat_annotations”,label = T) + NoLegend()
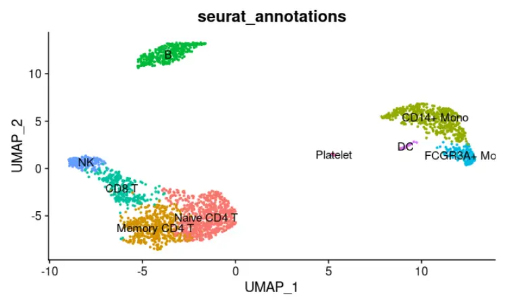
顺手设置一下数据集的ident
# set cluster to idents
Idents(pbmc3k.final) <- pbmc3k.final$seurat_annotations
3.加载R包
library(UCell)
library(irGSEA)
4.计算富集分数
当你的ncore设置大于1的时候,发生下面的错误:Error (Valid ‘mctype’: ‘snow’ or ‘doMC’),你应该检查一下你的AUCell 版本,确保版本大于等于1.14 。如果你比较懒,那你直接把ncore设置为1也是可以的,只是运行速度会稍微慢一点。
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = “RNA”,
slot = “data”, seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = “Homo sapiens”, category = “H”,
subcategory = NULL, geneid = “symbol”,
method = c(“AUCell”, “UCell”, “singscore”,
“ssgsea”),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = ‘Gaussian’)
# 返回一个Seurat对象,富集分数矩阵存放在RNA外的assay中
Seurat::Assays(pbmc3k.final)
5.整合差异基因集
为了评估基因集在某个细胞亚群中是否富集,通过多种基因集富集方法分别对单个细胞进行打分,并生成多个基因集富集分数矩阵。接着,通过wilcox检验计算各个基因集富集分数矩阵中每个细胞亚群差异表达的基因集。
单一的基因集富集分析方法不仅只能反映有限的信息,而且也容易带来误差。期待从多个角度解释复杂的生物学问题,并找到生物学问题中的共性部分。简单地为多种基因集富集分析方法的结果取共同交集,不仅容易得到少而保守的结果,而且忽略了富集分析方法中很多的其他信息,例如不同基因集的相对富集程度信息。
单纯地取共同交集无法体现基因集的相对富集程度,希望目标基因集在大部分富集分析方法中都是富集且富集程度没有明显差异。因此,通过RobustRankAggreg包中的秩聚合算法(robust rank aggregation, RRA)对差异分析的结果进行综合评估,筛选出在大部分基因集富集分析方法中都显著富集的基因集。
result.dge <- irGSEA.integrate(object = pbmc3k.final,
group.by = “seurat_annotations”,
metadata = NULL, col.name = NULL,
method = c(“AUCell”,”UCell”,”singscore”,
“ssgsea”))
class(result.dge)
6.可视化展示
我们内置了多种可视化的函数,不仅允许用户通过热图、气泡图、柱状图和upset图展示它们综合结果,而且允许用户通过密度散点图、半小提琴图、山峦图和密度热图展示目标基因集在具体富集分析方法中的表达水平和数据分布。其中,热图、upset图、密度热图主要由ComplexHeatmap包生成;气泡图和柱状图主要由ggplot2包、ggtree包、aplot包生成;密度散点图主要由Seurat包和Nebulosa包生成,半小提琴图主要由Seurat包和gghalves包生成;山峦图主要由Seurat包和ggridges包生成。最后,为了方便用户将可视化结果与其他的ggplot2对象进行拼图操作,我们也通过ggplotify包把输出结果转换为ggplot2对象。
作者:范垂钦_92be
链接:https://www.jianshu.com/p/463dd6e2986f
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
1). 全局展示
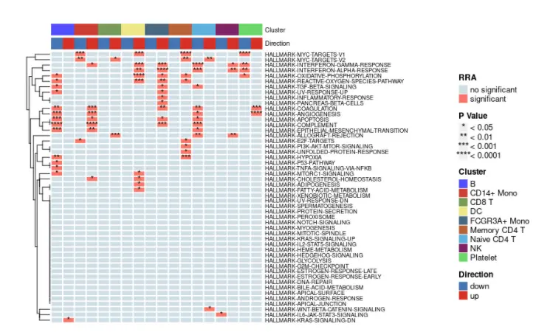
①.热图
热图展示了综合评价中具体基因集在每个细胞亚群是否具有统计学意义差异;
其中,浅蓝色的格子无统计学差异,红色的格子具有统计学差异。格子中的星号越多,格子的P值越小;
左边的聚类树代表不同基因集在不同细胞亚群中表达模式的相似性;
上方的条形图分别代表不同的细胞亚群,以及差异基因集在细胞亚群中是呈现上调还是下调趋势;
你还可以把method从’RRA”换成“ssgsea”,展示特定基因集富集分析方法中差异上调或差异下调的基因集;
irGSEA.heatmap.plot <- irGSEA.heatmap(object = result.dge,
method = “RRA”,
top = 50,
show.geneset = NULL)
irGSEA.heatmap.plot
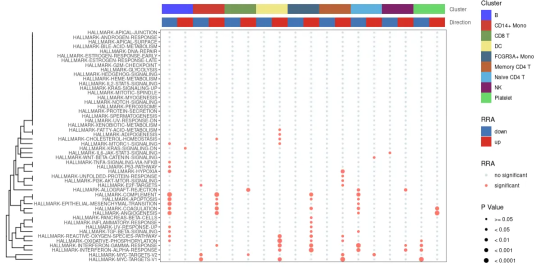
②.气泡图
气泡图展示了综合评价中具体基因集在每个细胞亚群是否具有统计学意义差异;
其中,浅蓝色的点无统计学差异,红色的点具有统计学差异。点越大,P值越小;
左边的聚类树代表不同基因集在不同细胞亚群中表达模式的相似性;
上方的条形图分别代表不同的细胞亚群,以及差异基因集在目标细胞亚群中是呈现上调还是下调趋势;
你还可以把method从’RRA”换成“ssgsea”,展示特定基因集富集分析方法中差异上调或差异下调的基因集;
如果运行中提示“ error (argument “caller_env” is missing, with no default)”,请卸载了ggtree包,并重新安装remotes::install_github(”YuLab-SMU/ggtree“)
irGSEA.bubble.plot <- irGSEA.bubble(object = result.dge,
method = “RRA”,
top = 50)
irGSEA.bubble.plot
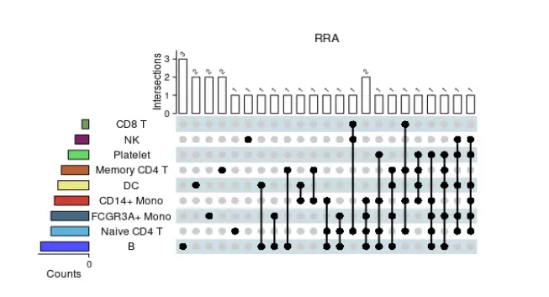
③.upset plot
upset图展示了综合评估中每个细胞亚群具有统计学意义差异的基因集的数目,以及不同细胞亚群之间具有交集的差异基因集数目;
左边不同颜色的条形图代表不同的细胞亚群;
上方的条形图代表具有交集的差异基因集的数目;
中间的气泡图单个点代表单个细胞亚群,多个点连线代表多个细胞亚群取交集.;
如果运行代码发生以下warning“the condition has length > 1 and
only the first element will be used”。这个警告可忽略。
irGSEA.upset.plot <- irGSEA.upset(object = result.dge,
method = “RRA”)
irGSEA.upset.plot
④.堆叠条形图
堆叠柱状图具体展示每种基因集富集分析方法中每种细胞亚群中上调、下调和没有统计学差异的基因集数目;
上方的条形代表每个亚群中不同方法中差异的基因数目,红色代表上调的差异基因集,蓝色代表下调的差异基因集;
中间的柱形图代表每个亚群中不同方法中上调、下调和没有统计学意义的基因集的比例;
irGSEA.barplot.plot <- irGSEA.barplot(object = result.dge,
method = c(“AUCell”, “UCell”, “singscore”,
“ssgsea”))
irGSEA.barplot.plot

2). 局部展示
①.密度散点图
密度散点图将基因集的富集分数和细胞亚群在低维空间的投影结合起来,展示了特定基因集在空间上的表达水平。其中,颜色越黄,代表富集分数越高;
scatterplot <- irGSEA.density.scatterplot(object = pbmc3k.final,
method = “UCell”,
show.geneset = “HALLMARK-INFLAMMATORY-RESPONSE”,
reduction = “umap”)
scatterplot

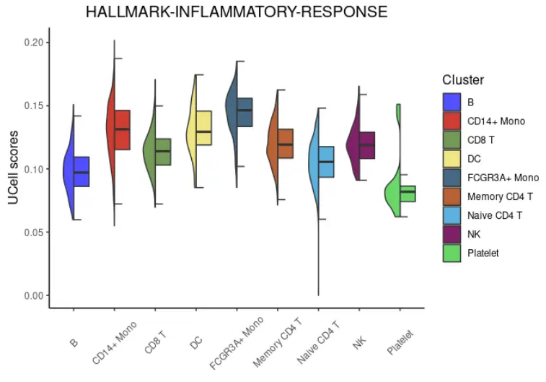
②.半小提琴图
半小提琴图同时以小提琴图(左边)和箱线图(右边)进行展示。不同颜色代表不同的细胞亚群;
halfvlnplot <- irGSEA.halfvlnplot(object = pbmc3k.final,
method = “UCell”,
show.geneset = “HALLMARK-INFLAMMATORY-RESPONSE”)
halfvlnplot
③.山峦图
山峦图中上方的核密度曲线展示了数据的主要分布,下方的条形编码图展示了细胞亚群具体的数量。不同颜色代表不同的细胞亚群,而横坐标代表不同的表达水平;
ridgeplot <- irGSEA.ridgeplot(object = pbmc3k.final,
method = “UCell”,
show.geneset = “HALLMARK-INFLAMMATORY-RESPONSE”)
ridgeplot

④.密度热图
密度热图展示了具体差异基因在不同细胞亚群中的表达和分布水平。颜色越红,代表富集分数越高;
densityheatmap <- irGSEA.densityheatmap(object = pbmc3k.final,
method = “UCell”,
show.geneset = “HALLMARK-INFLAMMATORY-RESPONSE”)
densityheatmap

小结
irGSEA 的 R 包合并所有六种方法的结果,通过采用稳健的 RRA 来确定目标基因集在不同评分方法中的统计显着性。irGSEA优先考虑用户友好性,允许直接输入表达式矩阵或与Seurat对象无缝交互。此外,irGSEA集成了几种用户友好的可视化工具来显示结果。是不是用起来方便多了?富集分析的结果也更准确了?感兴趣的小伙伴快来学习吧!大海的的分享到此结束啦!不过还是要给大家小送一波福利!大海哥给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!
参考文献
- Chuiqin Fan, Fuyi Chen, Yuanguo Chen, Liangping Huang, Manna Wang, Yulin Liu, Yu Wang, Huijie Guo, Nanpeng Zheng, Yanbing Liu, Hongwu Wang, Lian Ma, irGSEA: the integration of single-cell rank-based gene set enrichment analysis, Briefings in Bioinformatics, Volume 25, Issue 4, July 2024, bbae243, https://doi.org/10.1093/bib/bbae243