
大家好!今天我想和大家分享一个专门用于单细胞RNA测序数据标准化和质量控制的R包——SCONE包。通过使用SCONE包,我们可以系统化地处理单细胞RNA测序数据,揭示数据中的技术偏差和生物学差异,并进行详细的分析。除此之外,SCONE包还支持技术差异和批次效应的可视化分析以及漏检效应的评估,帮助我们深入理解和校正数据中的技术偏差。任何生信分析对生信高手小师妹来说都不是难题,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!
今天,小师妹将带领大家一起探索如何使用SCONE包来标准化和分析单细胞RNA测序数据。我们将学习如何安装SCONE包、加载数据、进行数据预处理,并分析和可视化技术差异和批次效应。通过今天的学习,同学们可以掌握使用SCONE包进行单细胞RNA测序数据分析的基本技能,并且能够将这些方法应用于自己的研究工作中,是不是已经迫不及待了,跟进小师妹的步伐,让我们一起开启对SCONE包的学习之旅吧!
本次介绍的R包需要较多的硬件资源,在服务器可以更加流畅运行,同学们如果没有自己的服务器欢迎联系我们进行服务器租赁~
SCONE包介绍
SCONE包(Single Cell Overview of Normalized Expression)是一个用于单细胞RNA测序数据标准化和质量控制的R包。通过系统化的分析流程,SCONE包帮助研究者揭示数据中的技术偏差和生物学差异。在实际应用中,SCONE包提供了多种标准化方法,并包含对技术差异和批次效应的可视化分析工具。例如,通过条形图可视化比对质量和总读取数,以评估各个批次间的差异。此外,SCONE包还包含漏检效应的评估功能,通过模型化假阴性率曲线,帮助研究者理解并校正数据中的技术偏差,确保数据分析的准确性。
SCONE包的安装
需要R语言版本为4.4,在控制台中输入以下命令:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager “)
BiocManager::install(“scone”) # 在BiocManager环境下安装SCONE
查看是否安装成功
packageVersion(“scone”) # 查看SCONE版本

显示为1.28.0版本,则表示已经安装了scone包。
除此之外,后续示例还需要使用scRNAseq包,我们可以提前安装,安装命令如下:
BiocManager::install(“scRNAseq”) # 在BiocManager环境下安装scRNAseq
packageVersion(“scRNAseq”) # 查看scRNAseq版本

数据载入和预处理
包和数据载入
我们将通过使用一个早期的scRNA-Seq数据集来演示基本的SCONE工作流。我们关注从四种生物条件中采样的65个人类细胞,分别为由多能干细胞衍生的培养神经前体细胞(“NPC”),在孕周16和21的初级皮质样本(分别为“GW16”和“GW21”),以及培养了3周的晚期皮质样本(“GW21+3”)。这些细胞的基因水平表达数据可以直接从Bioconductor上的scRNAseq包中加载。
首先我们需要载入SCONE包以及scRNAseq包
library(scone) #载入SCONE包
library(scRNAseq) #载入scRNAseq包
library(RColorBrewer) #载入RColorBrewer包
随后我们需要载入示例数据
fluidigm <- ReprocessedFluidigmData(assays = “rsem_counts”)
assay(fluidigm) <- as.matrix(assay(fluidigm)) #加载示例数据
数据预处理
在开始之前,我们将进行一些初步筛选,以去除低覆盖率重复样本和未检测到的基因特征,相关的代码如下:
# 初步样本筛选:仅高覆盖率
is_select = colData(fluidigm)$Coverage_Type == “High”
fluidigm = fluidigm[,is_select]
# 保留仅检测到的转录本
fluidigm = fluidigm[which(apply(assay(fluidigm) > 0,1,any)),]
使用SCONE包对单细胞数据进行评估
技术差异和批次效应的可视化分析
我们衡量比对质量的读数之一是与转录组对齐的读数的比例,我们可以使用简单的条形图来可视化该指标与生物批次的关系,代码如下:
# 定义颜色方案
cc <- c(brewer.pal(9, “Set1”))
# 每个生物学条件一个批次
batch = factor(colData(fluidigm)$Biological_Condition)
# 比对质量指标
qc = colData(fluidigm)[,metadata(fluidigm)$which_qc]
# 人类转录组映射读取比例的条形图
ralign = qc$RALIGN
o = order(ralign)[order(batch[order(ralign)])] # 按批次然后按值排序
barplot(ralign[o], col=cc[batch][o], border=cc[batch][o], main=”映射到人类转录组的读取百分比”)
legend(“bottomleft”, legend=levels(batch), fill=cc, cex=0.4)

我们可以看到批次之间存在适度差异,并且我们看到有一个GW21细胞的比对率相对于其他GW21批次特别低。这些类型的观察可以告知我们“质量较差”的文库或批次。我们同样也可以评估一下每个文库的读取数量,相关代码如下:
# 总读取数的条形图
nreads = qc$NREADS
o = order(nreads)[order(batch[order(nreads)])] # 按批次然后按值排序
barplot(nreads[o], col=cc[batch][o], border=cc[batch][o], main=”总读取数”)
legend(“topright”, legend=levels(batch), fill=cc, cex=0.4)
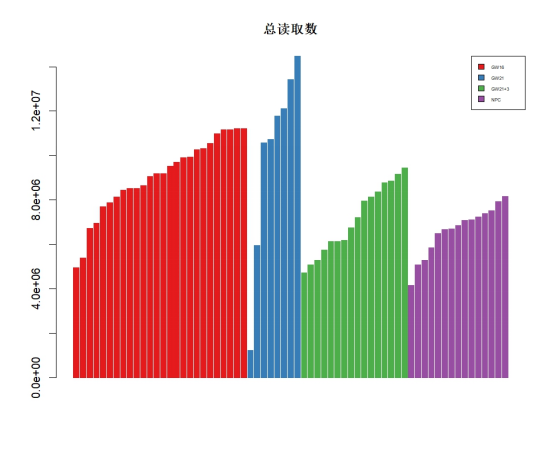
我们看到读取覆盖率在批次之间以及批次内部差异显著。这些覆盖率差异和其他技术特征可以导致对表达估计的非直观偏差。尽管一些偏差可以通过简单的文库大小归一化和细胞筛选来解决,但对多细胞数量的需求可能需要更复杂的归一化方法,以便比较多个批次的细胞。
漏检评估
单细胞特有的一个问题就是漏检效应。在建模漏检效应时面临的最大挑战之一是同时建模技术漏检和生物表达异质性。简化问题的一种方法是关注那些我们确信真正表达的基因。SCONE包包含了一些基因列表,这些基因被认为在所有人类组织中普遍且均匀地表达。如果我们假设这些基因在所有细胞中都真正表达,我们可以将所有零丰度观察标记为漏检事件。我们将检测失败建模为平均表达的逻辑函数,与该领域使用的标准逻辑模型保持一致,代码如下:
# 提取管家基因
data(housekeeping)
hk = intersect(housekeeping$V1, rownames(assay(fluidigm)))
# 平均log10(x+1)表达
mu_obs = rowMeans(log10(assay(fluidigm)[hk,]+1))
# 假定的假阴性
drop_outs = assay(fluidigm)[hk,] == 0
# 失败的逻辑回归模型
ref.glms = list()
for (si in 1:dim(drop_outs)[2]){
fit = glm(cbind(drop_outs[,si], 1 – drop_outs[,si]) ~ mu_obs,
family=binomial(logit))
ref.glms[[si]] = fit$coefficients
}
列表ref.glm包含了每个拟合的截距和斜率。我们现在可以可视化拟合曲线和相应的曲线下面积(AUC),代码如下:
par(mfrow=c(1,2))
# 绘制失败曲线并计算AUC
plot(NULL, main = “假阴性率曲线”,
ylim = c(0,1), xlim = c(0,6),
ylab = “失败概率”, xlab = “平均log10表达”)
x = (0:60)/10
AUC = NULL
for(si in 1:ncol(assay(fluidigm))){
y = 1/(exp(-ref.glms[[si]][1] – ref.glms[[si]][2] * x) + 1)
AUC[si] = sum(y)/10
lines(x, 1/(exp(-ref.glms[[si]][1] – ref.glms[[si]][2] * x) + 1),
type = ‘l’, lwd = 2, col = cc[batch][si])
}
# FNR AUC的条形图
o = order(AUC)[order(batch[order(AUC)])]
barplot(AUC[o], col=cc[batch][o], border=cc[batch][o], main=”FNR AUC”)
legend(“topright”, legend=levels(batch), fill=cc, cex=0.4)

由图可知,左图展示了不同细胞样本的假阴性率(漏检概率)随平均log10表达值的变化情况。曲线的横轴表示基因的平均log10表达值,纵轴表示漏检概率。不同颜色的曲线代表不同的生物条件。随着表达值的增加,漏检概率逐渐下降,这表明表达水平越高,基因被漏检的概率越低。右图展示了每个细胞样本的假阴性率曲线下面积(FNR AUC)的条形图。FNR AUC表示漏检率曲线下面的面积,是评价漏检效应的一个综合指标。条形图的颜色对应左图中的曲线颜色,表示不同的生物条件。我们可以看到,GW16(红色)和GW21(蓝色)样本的FNR AUC较高,表明这些样本的漏检效应较强。而GW21+3(绿色)和NPC(紫色)样本的FNR AUC较低,表明这些样本的漏检效应较弱。
以上就是对于SCONE包的全部介绍了,SCONE包提供了强大的工具和功能,可用于标准化和分析单细胞RNA测序数据及其在生物学研究中的应用。通过本文介绍的示例,我们展示了如何使用SCONE包来进行数据的预处理、技术差异和批次效应的可视化分析,以及漏检效应的评估,揭示了数据质量和生物条件之间的紧密联系。希望大家继续学习和探索SCONE包的更多功能,深入理解单细胞RNA测序数据,从而为单细胞转录组研究领域的进展做出更多贡献。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!