
今天大海哥要向大家介绍一个在基因组拷贝数变异分析领域中非常重要的R包——ACE包!ACE包是专门用于分析基因组拷贝数变异的强大工具。要知道,基因组拷贝数变异对于理解肿瘤的发生和发展至关重要,而ACE包提供了高效且准确的方法来进行这方面的分析。通过ACE包,我们可以构建拷贝数模型,评估样本中的细胞比例和拟合误差,并可视化基因组中的拷贝数变异情况。大海哥可是厉害的生信高手,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!
在本次学习中,我们不仅将学会如何安装和使用ACE包,还将掌握基因组拷贝数数据的预处理、模型构建、以及变异分析的方法。通过实际示例的演示,我们将更深入地理解基因组拷贝数变异的模式和影响,为我们解读肿瘤细胞的基因组特征提供重要线索。我相信,通过今天的学习,同学们都能够更加熟练地应用ACE包,为基因组变异分析的研究进步贡献自己的一份力量!接下来,就让大海哥带领大家正式开启对ACE包的学习之旅吧!
本次介绍的R包需要较多的硬件资源,在服务器可以更加流畅运行,同学们如果没有自己的服务器欢迎联系我们使用服务器租赁~
ACE包简介
ACE包是一个专门用于分析基因组拷贝数变异的R语言工具,适用于肿瘤研究等领域。通过ACE,用户可以构建拷贝数模型,评估样本中的细胞比例和拟合误差,并可视化基因组中的拷贝数变异情况。该工具提供了多种功能强大的函数和可视化工具,帮助研究人员深入理解肿瘤细胞中的拷贝数变异模式,识别潜在的基因扩增或缺失区域,并探索这些变异与肿瘤发展及治疗反应之间的关系。ACE的使用不仅简化了数据分析流程,也为基因组学研究提供了有力的工具支持。
ACE包安装
需要R语言版本为4.4,在控制台中输入以下命令:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager “)
BiocManager::install(“ACE”) # 在BiocManager环境下安装ACE
查看是否安装成功
packageVersion(“ACE”) # 查看ACE版本

显示为1.22.0版本,则表示已经安装了ACE包。
数据准备
想要使用ACE包分析细胞基因组拷贝数变异,我们首先需要载入需要用到的包和示例数据。在本文中,我们将使用ACE自带的2N肿瘤细胞数据作为我们的示例数据,相关的代码如下:
library(ACE) #载入ACE包
# 加载QDNAseq包中的示例数据集 “copyNumbersSegmented”
data(“copyNumbersSegmented”)
# 将加载的数据集赋值给变量 object
object <- copyNumbersSegmented
object # 打印对象 object 的内容和基本信息
# 显示对象的类型和主要属性,包括assayData、featureData和phenoData的简要概述
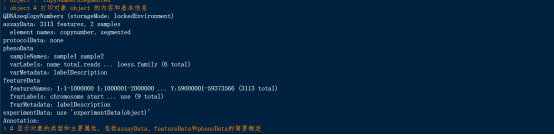
由上图可知,示例数据共包括2个数据样本,每个数据样本中有3113个特征基因,在本为中,我们只使用sample1作为示例数据,同学们如果有兴趣可以在完成本文学习之后自行分析sample2。
使用ACE包构建肿瘤细胞拷贝数模型
至此,我们已经载入了需要用到的包和数据,接下来我们可以直接使用ACE包中的singlemodel()函数,构建细胞的拷贝数模型,相关的代码如下:
model1 <- singlemodel(object, QDNAseqobjectsample = 1) # 创建单样本模型
bestfit1 <- model1$minima[tail(which(model1$rerror == min(model1$rerror)), 1)]
besterror1 <- min(model1$rerror) # 找到最佳拟合的拟合值和错误值
lastfit1 <- tail(model1$minima, 1)
lasterror1 <- tail(model1$rerror, 1) # 找到最后一个拟合的拟合值和错误值
# 绘制单样本的拷贝数数据图
singleplot(object, # 使用的数据对象
QDNAseqobjectsample = 1, # 指定的样本编号
cellularity = bestfit1, # 最佳拟合的细胞比例值
error = besterror1, # 最佳拟合的误差值
standard = model1$standard, # 模型的标准信息
title = “sample1 – binsize 1000 kbp – 379776 reads – 2N fit 1”)
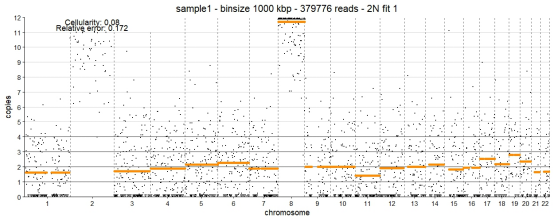
图为最佳拟合结果,在图中,X轴表示染色体位置,Y轴表示每个位置的拷贝数。每个黑色点表示一个千碱基对(kbp)片段的拷贝数值,橙色的水平线表示在这些位置的拷贝数平均值。可以看到,染色体8上有明显的拷贝数增加,达到11个拷贝,而在其他大部分染色体位置,拷贝数维持在2左右。这表明sample1中的某些区域存在显著的基因扩增现象,而其他区域则保持相对稳定。Cellularity为0.08,表示肿瘤细胞在样本中的比例相对较低,而Relative error为0.172,表示模型拟合的误差。这些信息表明,样本中的肿瘤细胞比例虽然较低,但染色体7的基因扩增非常显著,可能对肿瘤的发生和发展有重要作用。
# 绘制单样本的拷贝数数据图
singleplot(object, # 使用的数据对象
QDNAseqobjectsample = 1, # 指定的样本编号
cellularity = lastfit1, # 最后一个拟合的细胞比例值
error = lasterror1, # 最后一个拟合的误差值
standard = model1$standard, # 模型的标准信息
title = “sample1 – binsize 1000 kbp – 379776 reads – 2N fit 7”) # 图表的标题
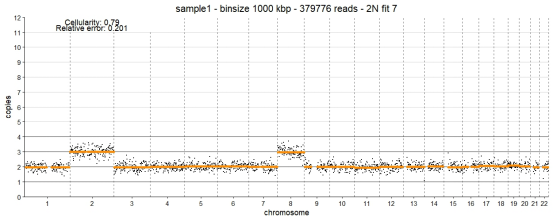
图中为对sample1进行最后拟合后的肿瘤细胞拷贝数变异分析结果,与最佳拟合不同,这次模型的拟合细胞比例为0.79,拟合误差为0.201。可以看到,染色体3和染色体8上有较为显著的拷贝数增加现象,这些区域的拷贝数均在2到4之间。相比最佳拟合,最后拟合显示的拷贝数变异更为复杂,可能由于在较高的细胞比例下,拷贝数变异区域增加,可能与肿瘤的恶性程度和发展速度有关。
以上就是对ACE包的全部介绍了。ACE包是一个专为肿瘤研究设计的工具,旨在分析基因组拷贝数变异。通过ACE包,我们能够构建拷贝数模型,评估样本中的细胞比例和拟合误差,并可视化基因组中的拷贝数变异情况。在今天的学习中,我们提供了ACE包的安装方法以及运行示例,展示了其在肿瘤细胞拷贝数变异分析方面的强大功能。希望同学们可以掌握ACE包的应用,让我们共同推动生命科学和医学的进步!
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!