前言
小伙伴们,还记不记得单细胞自动注释工具SingleR?是不是用起来爽爆了!让原本很难得细胞注释轻松秒杀!最近有小伙伴们问大海哥,当今空间转录组炙手可热,有没有类似的工具?小伙伴们真的快被人工标注折磨死了!虽然也有一些基于机器学习的方法已被广泛应用于识别空间点的群集,并使用标记基因解释其生物学身份。但这些方法通常因为缺乏与群集内已知结构建立明确联系的能力而受限。看到小伙伴们的困惑,大海哥连夜整理了份Pianno锦囊,给粉丝送福利!Pianno借鉴了计算机视觉中的「语义分割」思想,提出了「空间转录组语义注释」概念,并开发了空间转录组语义注释工具 Pianno,能够为组织内的空间点自动定义结构或细胞类型,从而结合来自多个维度的信息,加强对复杂生物系统的解释。下面跟着大海哥来学习吧!对了,有什么生信分析上的问题大家尽管咨询大海哥!没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找大海哥哦!练了十年生信分析的大海哥对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!
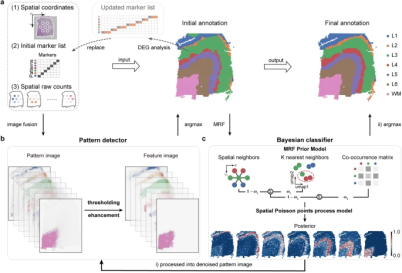
Pianno:创新的自动化空间转录组语义注释新工具
研究团队提出了一个基于贝叶斯框架的新工具 Pianno,该工具结合了马尔可夫随机场 (MRF) 与空间泊松点过程 (sPPP),充分利用了 sPPP 建模 RNA-seq 计数数据分布的能力,同时考虑了空间点的位置信息,可以使用预定义的标记基因列表,自动化地注释空间转录组数据中每个点的生物身份。
Pianno 提供两种更新注释的方法:
- 对于语义标注中的连续模式,建议将概率分布作为模式图像,返回给模式检测器 (Pattern detector) 进行更新标注;
- 对于分散或尖锐的图像模式,建议根据概率值直接更新标签,因为它可以保留详细信息。
总的来说,Pianno 简化了注释过程,同时采用启发式方法 (heuristic approach) 使用初始单个标记基因来识别额外的标记基因,可以最大限度减少对已知标记数量的输入。
生信数据处理起来占用内存实在太大了,放过自己的电脑吧!大海哥在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系大海哥,保证服务器的性价比最高哦!
代码实现
安装指南:从 PyPi 安装
# 创造一个Pianno环境
conda create -n Pianno python=3.9.10
# 激活环境
conda activate Pianno
# 在piano环境中安装R
conda install -c conda-forge r-base=4.1.1
#安装tensorflow和tensorflow-probability
#确保tensorflow2版本
pip install tensorflow-gpu==2.6.0
pip install tensorflow-probability==0.14.0
# install pianno from PyPi
pip install pianno
加载数据集
在这里,我们展示了人类背外侧前额叶皮层 (dlPFC) 数据集样本的注释过程。此演示演示了如何使用 Pianno 以 10x Visium 数据集为例进行语义注释。您可以扩展到其他平台生成的空间转录组数据或者自己的测序数据。
导入模块
import pianno as po
预处理
Step1:创建Pianno对象
Pianno 对象基于原始基因计数矩阵和空间斑点的坐标进行初始化。还进行预处理,包括尺寸系数的计算和质量控制。
data_path = “~/data/dlPFC-151674/”
count_file = “filtered_feature_bc_matrix.h5”
adata = po.CreatePiannoObject(data_path=data_path, count_file=count_file, min_spots_prop=0.01)
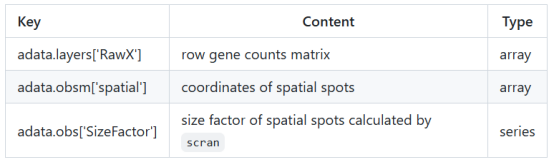
对于不是来自 10x Visium 的数据集,Pianno 对象也可以直接从逐个基因的原始计数矩阵 A 和空间坐标数据帧 B 构建:
adata = po.CreatePiannoObject(count_matrix=A, coordinates=B, min_spots_prop=0.01)
通过SAVER去除噪声
adata = po.SAVER(adata, layer_key=’DenoisedX’)
注意:短短一行代码大海哥运行了2小时 17分 23秒
这一步需要很长时间。我们提供了一个去噪对象,如果您不想等待,可以使用以下命令加载该对象:
adata = sc.read(“~/data/dlPFC-151674/adata.h5ad”)
您还可以选择其他降噪方法,只需将逐个基因去噪的基质存储到adata.layers[‘DenoisedX’] 。空间转录组数据的降噪对于注释是必要的。降噪的结果直接影响注释。
Step2:创建蒙版图像
二进制蒙版是通过在空间点和图像像素之间建立映射来创建的。
# 根据蒙版图像调整scale_factor的大小
adata = po.CreateMaskImage(adata, scale_factor=1)

此过程等效于对空间点进行分箱。对于空间坐标为数组状的数据集,比例因子可以设置为 1。对于空间坐标不是数组式的数据集,例如 SlidseqV2 和 stereo-seq,可以增加比例因子,直到输出合适的蒙版图像。
注释
研究中,为避免降噪、平滑、锐化等图像处理技术对生物原始特征造成破坏,研究团队基于原始计数构建了贝叶斯分类器 (Bayesian classifier) 以微调初始注释。同时,研究团队应用了高阶马尔科夫随机场 (Markov random field, MRF) 先验模型。在空间转录组学背景下,由于必须共同考虑每个位点的基因表达和空间位置,研究团队还采用了空间泊松点过程 (spatial Poisson point process, sPPP) 模型。
#设置数据和配置文件的存放路径。
sample_name = “GT151674”
config_path = “./Tutorials/” + sample_name
Step1:自动超参数选择
#为每个模式指定一个已知的标记基因
Patterndict = dict(L1 = [‘CXCL14’],
L2 = [‘HPCAL1’],
L3 = [‘CALB1’],
L4 = [‘NEFH’],
L5 = [‘PCP4’],
L6 = [‘KRT17’],
WM = [‘MOBP’])
如果连接失败,请再尝试几次。
打开Web UI url以可视化超参数调优过程。
默认实验最多持续10分钟,
可根据实际情况进行修改。
adata = po.AutoPatternRecognition(adata,
Patterndict=Patterndict,
config_path=config_path,
param_tuning=True,
max_experiment_duration=’10m’)

#输出上一步保存的最优参数。
with open(join(config_path, “best_params.json”),’r’) as f:
best_params_dict = json.load(f)
for key in best_params_dict:
best_params = best_params_dict[key]
best_params
Step2:标记选择
将前10个deg作为候选标记基因,提出一种模式预测
Patterndict = po.ProposedPatterndict(adata, top_n=10)
# 候选标记基因的可视化
for k, v in Patterndict.items():
print(k)
print(v)
with mpl.rc_context({‘axes.facecolor’: ‘black’,
‘figure.figsize’: [4.5, 5]}):
sc.pl.spatial(adata, #cmap=’magma’,
layer=’DenoisedX’,
color=v,
ncols=5, size=5,
spot_size=25,
vmin=0, vmax=’p99′
)
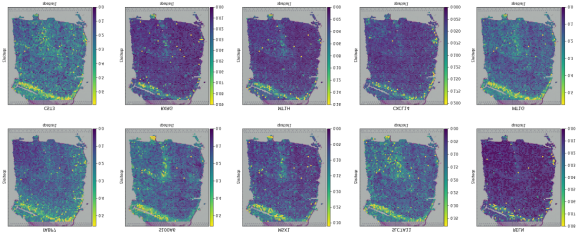
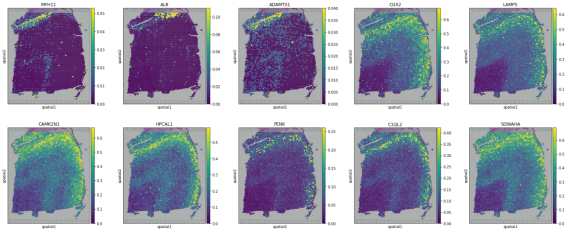
Step3:初始模式识别
通过为每个模式选择1-3个高质量基因来构建标记列表,并从上述候选标记基因。
Patterndict = dict(L1 = [‘CXCL14′,’MT1G’,’FABP7′],
L2 = [‘HPCAL1′,’C1QL2’],
L3 = [‘CALB1′,’LINC01007’],
L4 = [‘NEFH’,’SYT2′,’NEFM’],
L5 = [‘PCP4′,’TMSB10’],
L6 = [‘KRT17’],
WM = [‘MOBP’,’MBP’])
Patterndict也可以是一个整数N。piano将选择前N个候选标记基因来构建标记列表。
adata = po.AutoPatternRecognition(adata,
Patterndict=Patterndict,
config_path=config_path,
param_tuning=False)

Step4:注释改进
adata = po.AnnotationImprovement(adata)

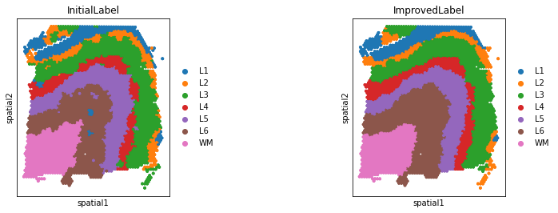
小结
Pianno工具为自动化空间转录组数据的生物身份注释提供了一种创新解决方案。Pianno的应用不仅展示了在人脑皮质层结构和肿瘤微环境样本中识别复杂生物模式的能力,而且还证明了其在提高注释准确性和处理效率方面的显著优势。这一成就为深入理解复杂生物系统中的基因表达模式开辟了新的途径,预示着Pianno在促进生物医学研究和新治疗策略发现方面的巨大潜力。随着Pianno在更多数据集上的应用和验证,其注释能力和准确性预计将得到进一步提升,为揭示组织内细胞间相互作用和基因表达模式的复杂网络提供强有力的支持。最后大海哥给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!