
同学们,今天大海哥要向大家介绍一个在分子生物学领域中非常重要的软件——BLAST+!BLAST+ 是一款用于序列比对的强大工具。要知道,在生物序列中,比较不同序列之间的相似性可以帮助我们揭示它们的进化关系、功能特性和潜在的生物学意义。通过 BLAST+,我们可以快速、高效地比对 DNA、RNA 和蛋白质序列,发现同源基因,进行功能注释,以及探索进化过程中的关键变化。序列比对对大海哥来说根本不是难题,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!
在本次学习中,我们不仅将学会如何安装和使用 BLAST+,还将掌握对序列数据进行比对和分析的方法。通过实际示例的演示,我们将更深入地理解序列相似性的计算原理和比对结果的解读技巧。BLAST+ 不仅可以帮助我们识别已知的基因和功能元件,还可以发现潜在的新基因和变异,为基因功能研究和生物学探索提供了重要支持。通过今天的学习,同学们将能够更加熟练地应用 BLAST+,从而在基因组学、功能基因组学和进化生物学的研究中取得更大的进展。接下来,同学们跟随大海哥的步伐,让我们正式开启对 BLAST+ 的学习之旅吧!
本次介绍的工具需要在服务器上才能正常运行,同学们如果没有自己的服务器欢迎联系我们进行服务器租赁~
BLAST+软件介绍
BLAST+ 是一套用于序列比对的软件套件,其中包括了多种版本的 BLAST 程序,如 blastp 用于蛋白质序列比对。通过计算得分和期望值(E-value),blastp 能有效地比对蛋白质序列,找出相似性和同源性。用户可以通过调整参数优化比对结果,如设置阈值、选择合适的序列数据库。BLAST+ 提供了高效的并行计算支持,适用于广泛的生物信息学研究,从基础的序列相似性分析到复杂的功能注释和进化研究。
BLAST+软件安装
BLAST+必须要在服务器上运行,需要同学们有一些Linux系统的基础知识,如果没有也不用担心,大海哥带你一步一步来,跟紧大海哥的步伐,让我们从BLAST+的安装开始,慢慢学习如何使用这款强大的工具吧。
需要的系统:Linux系统,需要的软件支持:conda
为了避免我们现在的系统环境不符合BLAST+软件的要求,所以我们需要为BLAST+安装一个虚拟的工作环境,在不更改现在系统环境的前提下,安装BLAST+,命令如下:
conda create –name blast+ # 创建一个BLAST+环境

遇到图上提示,输入y即可
创建完环境后,我们激活环境,命令如下:
conda activate blast + # 激活BLAST+环境
显示(blast+)则表明我们已经成功创建并且激活了BLAST+环境,接着我们就可以在该环境下安装BLAST+了,命令如下:
conda install blast # 在conda环境中安装blast
遇到图上提示,输入y即可
耐心等待安装完成后,我们可以输入以下命令测试是否安装成功。
blastp -help #唤醒BLAST+ 参考手册
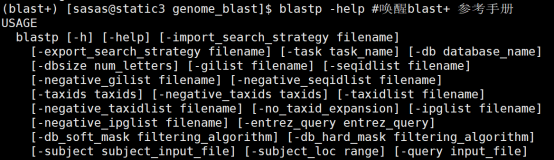
如果显示如图所示的命令参数提示,就表明已经成功安装了BLAST+程序。
使用BLAST+进行蛋白质序列比对
BLAST+主要命令
BLAST+ 是 NCBI 提供的一组用于序列比对和分析的工具集,包括多种命令和程序,每个命令用于不同类型的序列比对。以下是 BLAST+ 中一些主要的命令及其简要介绍:
blastp:用于比对蛋白质序列与蛋白质数据库。
blastn:用于比对核酸序列与核酸数据库。
blastx:用于将未知的核酸序列翻译成氨基酸序列,然后与蛋白质数据库比对。
tblastn:用于将蛋白质序列翻译成核酸序列,然后与核酸数据库比对。
tblastx:用于比对核酸序列的六种翻译框(三种正向加三种反向)与核酸数据库的六种翻译框。
makeblastdb:用于创建 BLAST 数据库。
使用blastp进行蛋白质序列比对
在本文中,大海哥主要向大家介绍一下blastp命令的语法,我们将对从Uniport获取的人体和大鼠的血红蛋白β亚基(Hemoglobin subunit beta, HBB)蛋白质序列进行比对。
首先我们需要从Uniport上下载人体和大鼠的血红蛋白β亚基的氨基酸序列,命令如下:
wget -c https://rest.uniprot.org/uniprotkb/P68871.fasta #使用wget命令下载人体血红蛋白β亚基的氨基酸序列
wget -c https://rest.uniprot.org/uniprotkb/P11517.fasta #使用wget命令下大鼠血红蛋白β亚基的氨基酸序列
mv P68871.fasta ./Human.fasta #更改文件名称
mv P11517.fasta ./Rat.fasta #更改文件名称
ls #查看下载的文件
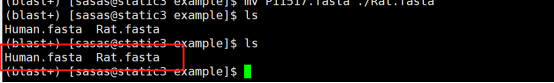
显示如上图,则表示我们已经成功下载了人体和大鼠的血红蛋白β亚基的氨基酸序列
接着,我们使用blastp命令进行蛋白质系列比对,命令如下:
makeblastdb -in Rat.fasta -dbtype prot -out Rat_db
# 创建 BLAST 数据库 -in Rat.fasta:指定输入的 FASTA 格式的文件(这里是大鼠的序列)-dbtype prot:指定数据库类型为蛋白质 -out Rat_db:指定输出的数据库名称为 Rat_db
blastp -query Human.fasta -db Rat_db -out results.txt
# 运行 BLASTP 进行蛋白质序列比对 -query Human.fasta:指定查询序列文件(这里是人类的序列)-db Rat_db:指定要比对的数据库名称(这里是刚创建的大鼠数据库)-out results.txt:指定输出文件名称为 results.txt
蛋白质序列比对结果查看和解析
以上命令运行结束后,会在当前目录输出一个results.txt文件,即比对的结果,我们可以使用cat命令查看。
cat results.txt #查看比对结果
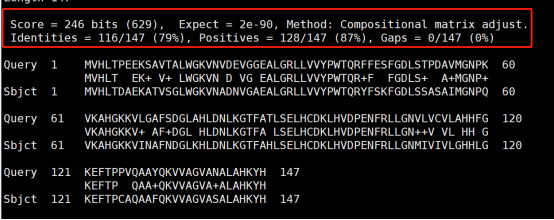
显示如上图,其中
Score (246 bits / 629):比对得分表示查询序列与目标序列的匹配质量。246 bits 是标准化后的分数,629 是原始得分。高分数表示比对质量好。
Expect (2e-90):期望值表示在随机序列中出现这种匹配的概率。2e-90 表示概率非常低,表明比对结果非常显著。
Identities (116/147, 79%):在147个比对位置中,有116个位置的残基完全匹配,占比79%。这表示查询序列和目标序列在这些位置是完全相同的。
Positives (128/147, 87%):在147个比对位置中,有128个位置的残基要么完全匹配,要么是保守替换,占比87%。保守替换表示残基虽然不同,但具有相似的化学性质。
Gaps (0/147, 0%):在147个比对位置中,没有插入或缺失情况,表示两个序列在比对过程中没有间隙。
总体而言,这些结果表明人体和大鼠的血红蛋白β亚基的氨基酸序列在结构和功能上可能具有高度的相似性。
以上就是对于 BLAST+ 软件的全部介绍了。通过本文,我们了解了 BLAST+ 作为强大的序列比对工具的功能和应用场景。BLAST+ 结合了灵活性和高效性,是生物信息学研究中不可或缺的利器。希望同学们能够继续深入学习和探索 BLAST+,充分利用这一工具在各自的研究领域中挖掘更多的科学发现。通过不断实践和应用,大家一定能在分子生物学和进化研究中取得更大的进步和成果。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!