前言
生存分析文章中比比皆是,你的建模方法用对了吗?你的模型真的准确吗?是不是东拼西凑找来代码,可视化之后草草了事?大海哥最经听到大家说生存分析建模实在是太复杂了,对于建模了解的太肤浅,东拼西凑才能完成生存分析全部流程!看到大家的呼声,大海哥赶紧放下手里的实验,连夜给大家整理了一份生存分析锦囊。从数据准备到最终校准模型的完整工作流程大海哥都给大家准备好了!对于许多疾病来说,目标事件的发生时间(例如死亡时间或疾病进展)是一个广泛使用的终点和患者预后,因此,识别基因组、分子和临床标志物以预测患有癌症等复杂疾病的患者的生存或进展已经变得很流行。大海哥给大家提供了一份锦囊用于使用组学和标准临床数据进行生存分析,特别关注生存相关组学特征的特征选择和生存模型验证,还涵盖了许多用于特征选择和生存预测的惩罚回归和贝叶斯模型,并考虑了它们的具体假设和应用。大海哥就是这么的宠粉,有什么生信分析上的问题大家尽管咨询大海哥!没有时间学习的小伙伴们也不要着急哦!有需要生信分析的小伙伴们也可以找大海哥哦!练了十年生信分析的大海哥对于生信分析知识已经如鱼得水从分析到可视化直到你满意为止!
生信数据处理起来占用内存实在太大了,放过自己的电脑吧!大海哥在这里给大家送上福利了,有需要服务器的小伙伴们,欢迎大家联系大海哥,保证服务器的性价比最高哦!
代码教程
小伙伴们注意啦!由于分享篇幅有限,更详细的建模流程以及代码私信大海哥获取哦!
我们将使用TCGA数据来证明:
- 不同的数据类型
- 生存和组学数据的预处理
- 通过经典统计方法分析生存数据
- 组学数据的无监督学习
- 生存和组学数据的频率和贝叶斯监督学习
首先,我们加载本教程中使用的所有必要库。然后,我们使用TCGAbiolinks软件包中的函数查询和下载来自多种癌症类型的TCGA生存和临床数据。输入数据直接通过R包TCGAbiolinks下载哦!
1.TCGA临床和生存数据
#加载所有的R包
library(“TCGAbiolinks”)
library(“SummarizedExperiment”)
library(“DESeq2”)
library(“M3C”)
library(“dplyr”)
library(“ggplot2”)
library(“survival”)
library(“survminer”)
library(“glmnet”)
library(“plotmo”)
library(“grpreg”)
library(“SGL”)
library(“psbcGroup”)
library(“psbcSpeedUp”)
library(“BhGLM”)
library(“risksetROC”)
library(“riskRegression”)
library(“peperr”)
library(“c060”)
library(“rms”)
library(“survAUC”)
library(“regplot”)
#下载临床数据,使用GDC api方法提取多种肿瘤的数据
cancer_types <- c(
“TCGA-BLCA”, “TCGA-BRCA”, “TCGA-COAD”, “TCGA-LIHC”,
“TCGA-LUAD”, “TCGA-PAAD”, “TCGA-PRAD”, “TCGA-THCA”
)
clin <- NULL
for (i in seq_along(cancer_types)) {
tmp <- TCGAbiolinks::GDCquery_clinic(project = cancer_types[i], type = “clinical”)
clin <- rbind(clin, tmp[, c(
“project”, “submitter_id”, “vital_status”,
“days_to_last_follow_up”, “days_to_death”,
“age_at_diagnosis”, “gender”, “race”,
“ethnicity”, “ajcc_pathologic_t”
)])
}
#提取每位患者的观察时间,以年为单位
clin$time <- apply(clin[, c(“days_to_death”, “days_to_last_follow_up”)], 1, max, na.rm = TRUE) / 365.25
clin$age <- clin$age_at_diagnosis / 365.25
clin$status <- clin$vital_status
clin <- clin[, c(“project”, “submitter_id”, “status”, “time”, “gender”, “age”, “race”, “ethnicity”)]
# extract patients with positive overall survival time
clin <- clin[(clin$time > 0) & (clin$status %in% c(“Alive”, “Dead”)), ]
# 患者状况、性别和种族的频率表
clin %>%
dplyr::count(status, gender, ethnicity) %>%
group_by(status) %>%
mutate(prop = prop.table(n))
# 按癌症类型筛选
ID <- 1:nrow(clin)
clin %>%
ggplot(
aes(y = ID, x = time, colour = project, shape = factor(status))
) +
geom_segment(aes(x = time, y = ID, xend = 0, yend = ID)) +
geom_point() +
ggtitle(“”) +
labs(x = “Years”, y = “Patients”) +
scale_shape_discrete(name = “Status”, labels = c(“Censored”, “Dead”)) +
scale_color_discrete(
name = “Cancer”,
labels = c(“Bladder”, “Breast”, “Colon”, “Liver”, “Lung adeno”, “Pancreatic”, “Prostate”, “Thyroid”)
) +
theme(legend.position = “top”, legend.direction = “vertical”) +
guides(color = guide_legend(nrow = 2, byrow = TRUE))
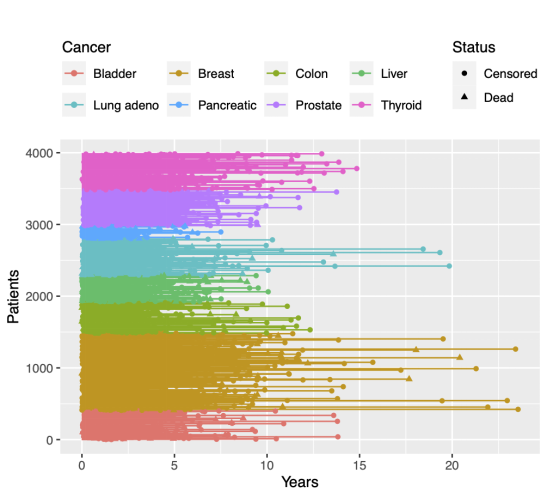
TCGA泛癌患者的总生存时间和状况
我们使用函数来查询和使用以及下载一种癌症类型(乳腺癌)的TCGA组学数据。函数中的参数指定组学数据的类型。请注意,下载的组学数据伴随着元数据,包括生存结果、临床和人口统计学变量。
# 使用GDC api方法下载TCGA乳腺癌(BRCA) mRNA-Seq数据
query <- TCGAbiolinks::GDCquery(
project = “TCGA-BRCA”,
data.category = “Transcriptome Profiling”,
data.type = “Gene Expression Quantification”,
workflow.type = “STAR – Counts”,
experimental.strategy = “RNA-Seq”,
sample.type = c(“Primary Tumor”)
)
TCGAbiolinks::GDCdownload(query = query, method = “api”)
dat <- TCGAbiolinks::GDCprepare(query = query)
SummarizedExperiment::assays(dat)$unstranded[1:5, 1:2]
建议先对RNA-seq数据采用DESeq2或TMM归一化方法,再进行进一步统计分析。在这里,我们演示了如何使用R/Bioconductor软件包DESeq2来规范化RNA计数数据。
meta <- colData(dat)[, c(“project_id”, “submitter_id”, “age_at_diagnosis”, “ethnicity”, “gender”, “days_to_death”, “days_to_last_follow_up”, “vital_status”, “paper_BRCA_Subtype_PAM50”, “treatments”)]
meta$treatments <- unlist(lapply(meta$treatments, function(xx) {
any(xx$treatment_or_therapy == “yes”)
}))
dds <- DESeq2::DESeqDataSetFromMatrix(assays(dat)$unstranded, colData = meta, design = ~1)
dds2 <- DESeq2::estimateSizeFactors(dds)
RNA_count <- DESeq2::counts(dds2, normalized = TRUE)
RNA_count[1:5, 1:2]
为了同时使用临床/人口学变量和组学数据进行生存分析,在以下代码中,我们提取了女性乳腺癌患者及其相应的生存结果、临床/人口学变量和RNA-seq特征。
meta$time <- apply(meta[, c(“days_to_death”, “days_to_last_follow_up”)], 1, max, na.rm = TRUE) / 365.25
meta$status <- meta$vital_status
meta$age <- meta$age_at_diagnosis / 365.25
clin <- subset(meta, gender == “female” & !duplicated(submitter_id) & time > 0 & !is.na(age))
clin <- clin[order(clin$submitter_id), ]
RNA_count <- RNA_count[, rownames(clin)]
2.生存分析
对于我们在上一节中提取的TCGA乳腺癌患者的数据,可以通过survival包中的函数获得生存概率的Kaplan-Meier估计。
# KM评估
clin$status[clin$status == “Dead”] <- 1
clin$status[clin$status == “Alive”] <- 0
clin$status <- as.numeric(clin$status)
sfit <- survival::survfit(Surv(time, status) ~ 1, data = clin)
# 计算1、3、5年时间点的生存率
summary(sfit, times = c(1, 3, 5))
theme_set(theme_bw())
ggsurv <- survminer::ggsurvplot(sfit,
conf.int = TRUE, risk.table = TRUE,
xlab = “Time since diagnosis (year)”,
legend = “none”, surv.median.line = “hv”
)
ggsurv$plot <- ggsurv$plot + annotate(“text”, x = 20, y = 0.9, label = “+ Censor”)
ggsurv
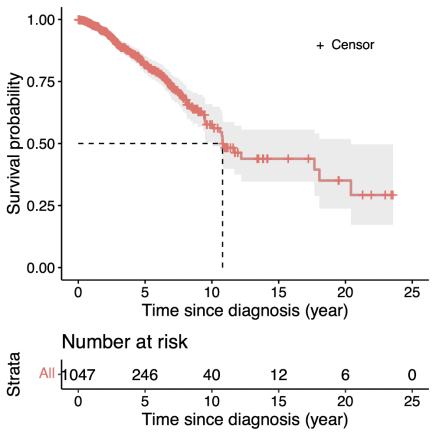
来自 TCGA 的 1061 例 BRCA 患者数据的 Kaplan-Meier 曲线。从生存曲线中可以揭示一些基本的统计数据。例如,所有乳腺癌患者的估计中位生存时间,即生存概率为50%的时间为10.8年(图中的虚线),1年、5年和10年生存概率分别为0.988、0.853和0.658。
对数秩检验可用于测试两组患者 [例如接受治疗(药物/放射治疗)或未治疗] 是否具有相同(原假设)或不同的生存函数(替代假设),并提供相应的 P 值。log-rank 检验还可用于根据其他分类变量比较任何患者亚组的生存概率。
为了比较两组患者的生存曲线,例如治疗(即药物或放射治疗)或非治疗,该函数可以执行对数秩检验来比较两条生存曲线。或者,使用包含治疗组作为协变量的公式的函数可以返回每个组的 (KM) 生存概率。然后,带有对象的函数将绘制两条生存曲线并执行对数秩测试,如下图所示。
survival::survdiff(Surv(time, status) ~ treatments, data = clin)
sfit2 <- survfit(Surv(time, status) ~ treatments, data = clin)
ggsurv <- ggsurvplot(sfit2,
conf.int = TRUE, risk.table = TRUE,
xlab = “Time since diagnosis (year)”, legend = c(.6, .9),
legend.labs = c(“No”, “Yes”), legend.title = “Treatment”,
risk.table.y.text.col = TRUE, risk.table.y.text = FALSE
)
ggsurv$plot <- ggsurv$plot +
annotate(“text”, x = 21, y = 1, label = “+ Censor”) +
annotate(“text”, x = 22, y = .88, label = paste0(“Log-rank test:\n”, surv_pvalue(sfit2)$pval.txt))
ggsurv
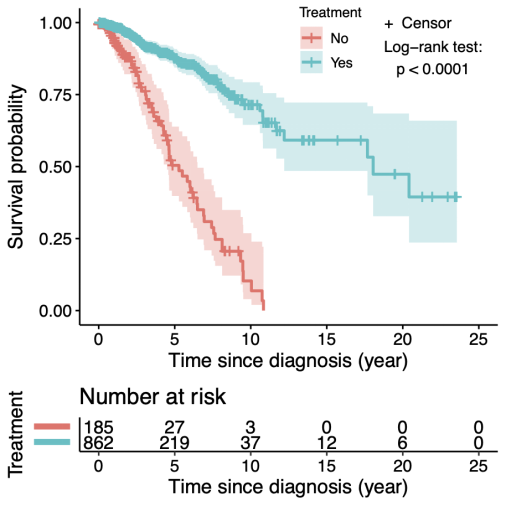
来自TCGA的BRCA患者生存数据的Kaplan-Meier曲线按治疗(即药物或放射治疗)或非治疗分组。log-rank 检验是比较对应于两组患者的两种生存分布。
为了分析连续变量(例如年龄)是否与生存结果相关,我们可以使用函数来拟合 Cox 模型,该函数类似于拟合线性模型的函数。
fit_cox <- coxph(Surv(time, status) ~ age, data = clin)
summary(fit_cox)
Cox 模型假设协变量具有比例风险和对数线性。为了检查临床或人口统计变量(例如年龄)的对数线性,我们可以拟合年龄效应的惩罚平滑样条。下面的代码显示,平滑样条的非线性部分具有显著的影响。因此,年龄的对数线性假设不满足。
fit_cox_spline <- coxph(Surv(time, status) ~ pspline(age), data = clin)
fit_cox_spline
为了检查年龄的比例风险,我们可以添加一个随时间变化的协变量。
survival::cox.zph(fit_cox, transform = “log”)
在这里,检查对数线性或比例风险的方法只能用于低维数据设置。当包括高维组学数据时,目前没有检查对数线性或比例风险的标准方法。
3. 生存模型验证
预后模型的理想评估是基于完全独立的验证数据,因为基于训练数据构建的高维生存模型可能会过拟合。如果没有独立的验证数据,建议使用基于重采样的方法估计模型预测性能的不确定性。例如,可以通过反复将数据集拆分为训练/验证集并使用各种评估指标评估模型在不同验证集上的性能来完成此操作。
经典模型评估
为了评估统计模型的性能,我们首先将数据拆分为训练数据集和验证数据集。例如,我们可以将 TCGA 的 1047 名 BRCA 患者随机拆分为80%作为训练集和20%作为验证集。
set.seed(123)
n <- nrow(x)
idx <- sample(1:n, n * 0.8, replace = FALSE)
x_train <- x[idx, ]
y_train <- y[idx, ]
x_validate <- x[-idx, ]
y_validate <- y[-idx, ]
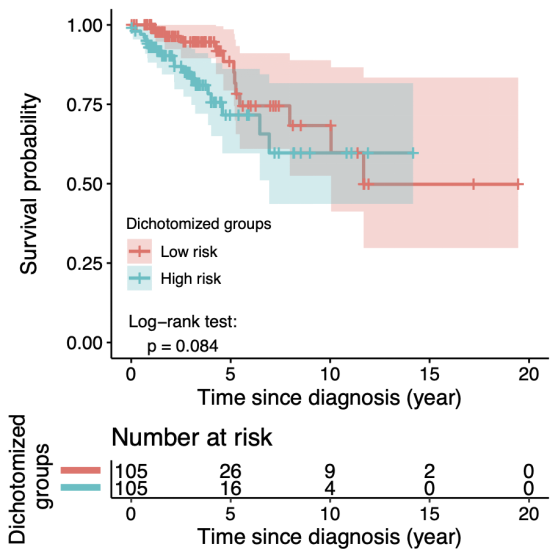
证明生存模型预后能力的最简单方法是对预后评分(即线性预测因子)进行二分法LP在Cox模型中的中值,然后使用对数秩检验来比较两组患者的生存曲线。
# 训练Lasso Cox模型,类似于其他Cox类型模型
set.seed(123)
cvfit <- cv.glmnet(x_train, y_train, family = “cox”, nfolds = 5, penalty.factor = pf)
pred_lp <- predict(cvfit, newx = x_validate, s = cvfit$lambda.min, type = “link”)
# 根据预后评分(线性预测因子)中位数进行二分类,将验证患者分为两组
group_dichotomize <- as.numeric(pred_lp > median(pred_lp))
# 根据KM估计绘制两条生存曲线,并通过log-rank检验进行比较
dat_tmp <- data.frame(time = y_validate[, 1], status = y_validate[, 2], group = group_dichotomize)
sfit <- survfit(Surv(time, status) ~ group, data = dat_tmp)
ggsurv <- ggsurvplot(sfit,
conf.int = TRUE, risk.table = TRUE,
xlab = “Time since diagnosis (year)”, legend = c(.2, .3),
legend.labs = c(“Low risk”, “High risk”), legend.title = “Dichotomized groups”,
risk.table.y.text.col = TRUE, risk.table.y.text = FALSE
)
ggsurv$plot <- ggsurv$plot +
annotate(“text”, x = 2.6, y = .03, label = paste0(“Log-rank test:\n”, surv_pvalue(sfit)$pval.txt))
ggsurv$table <- ggsurv$table + labs(y = “Dichotomized\n groups”)
ggsurv
预后评分也可以根据分位数分为三组或更多组,并使用log-rank检验来比较多条生存曲线的差异。
group <- pred_lp
group[pred_lp >= quantile(pred_lp, 2 / 3)] <- 3
group[pred_lp >= quantile(pred_lp, 1 / 3) & pred_lp < quantile(pred_lp, 2 / 3)] <- 2
group[pred_lp < quantile(pred_lp, 1 / 3)] <- 1
#根据KM估计绘制两条生存曲线,并通过log-rank检验进行比较
dat_tmp <- data.frame(time = y_validate[, 1], status = y_validate[, 2], group = group)
sfit <- survfit(Surv(time, status) ~ group, data = dat_tmp)
ggsurv <- ggsurvplot(sfit,
conf.int = TRUE, risk.table = TRUE,
xlab = “Time since diagnosis (year)”, legend = c(.2, .3),
legend.labs = c(“Low risk”, “Middle risk”, “High risk”), legend.title = “Groups”,
risk.table.y.text.col = TRUE, risk.table.y.text = FALSE
)
ggsurv$plot <- ggsurv$plot +
annotate(“text”, x = 3.5, y = .05, label = paste0(“Log-rank test:\n”, surv_pvalue(sfit)$pval.txt))
ggsurv
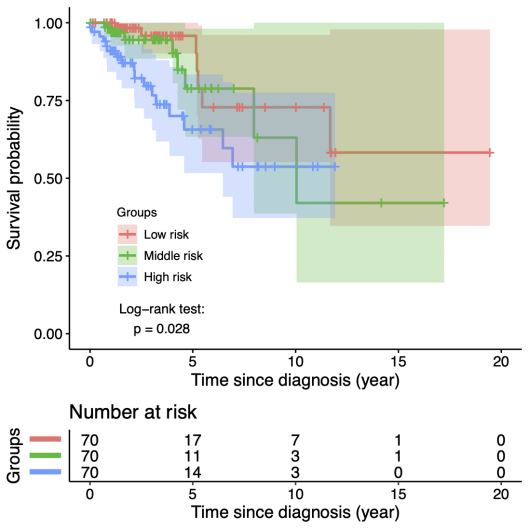
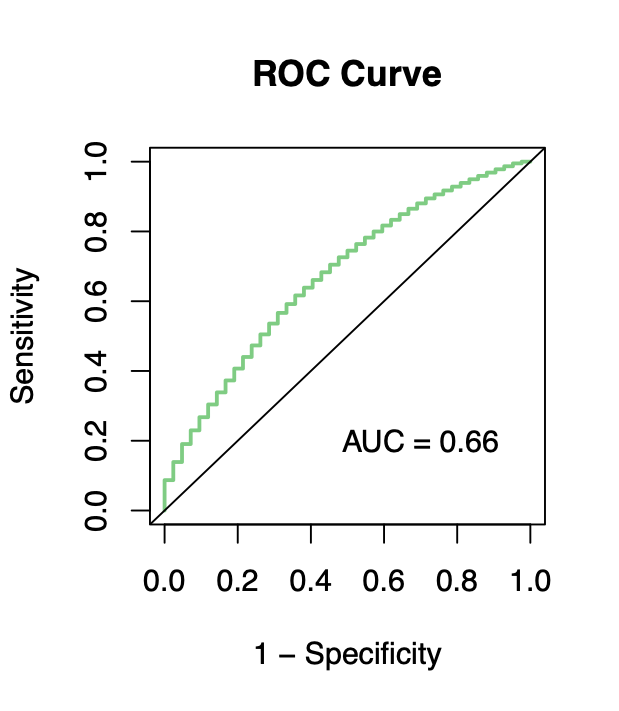
#ROC曲线
ROC <- risksetROC(
Stime = y_validate[, 1], status = y_validate[, 2],
marker = pred_lp, predict.time = 5, method = “Cox”,
main = “ROC Curve”, col = “seagreen3”, type = “s”,
lwd = 2, xlab = “1 – Specificity”, ylab = “Sensitivity”
)
text(0.7, 0.2, paste(“AUC =”, round(ROC$AUC, 3)))
小结
大海哥带大家学习了一个生存分析的一般工作流程,该工作流程适用于高维组学数据,作为识别生存相关特征和验证生存模型时的输入。特别是,我们重点关注了生存分析中常用的Cox型惩罚回归和分层贝叶斯模型,这些模型对于高维数据特别有用。最后大海哥给大家介绍一个云工具!同学们如果觉得自己的代码水平一般,对于很多的参数不知道怎么改,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表,字体大小、标题等也可一键更改。感兴趣的小伙伴去云生信(http://www.biocloudservice.com/home.html)体验一下吧!