大家好!我是小果!小伙伴们,是不是在进行生物数据分析时,经常因为序列比对的问题而感到苦恼呢?复杂的序列数据、繁琐的比对过程,总是让我们感到力不从心。今天小,果将带大家用Biostrings R包的函数来解决这个难题,掌握本期内容,序列比对的问题都将迎刃而解!Biostrings R包提供了非常强大的序列比对功能,不仅支持多样化的比对算法,还能轻松实现比对结果的可视化。小伙伴们无需再为复杂的比对过程而烦恼,只需几个简单的函数调用,就能轻松完成序列比对任务。快来体验Biostrings R包的强大魅力吧,让我们一起告别序列比对的烦恼,轻松探索生物数据的奥秘!同时呢,十年的生信之路,小果已经练就了一身扎实的本领,现在准备用这身本事为小伙伴们服务啦!如果你在生信分析上遇到了难题,那就来找小果吧!小果会用自己的专业知识和技能,为你解决困扰,助你一臂之力。期待你的联系哦~
一、 用到的包简要介绍
- Biostrings R包
Biostrings,它作为R语言中的一个核心生物信息学包,为处理和分析生物序列数据提供了强有力的支持。Biostrings的核心功能之一是处理各种生物序列数据,包括DNA、RNA和蛋白质序列。它提供了丰富的函数来读取、编辑、合并和转换这些序列,让我们能够轻松地对序列数据进行预处理和整理。除了序列处理功能外,Biostrings还具备强大的序列比对能力。它提供了多种比对算法,包括全局比对、局部比对和半全局比对等,以满足不同研究需求。我们还可以使用Biostrings中的函数来搜索特定序列在基因组中的位置,并生成相应的可视化图表。
- ggplot2 R包
ggplot2,我们的老朋友~非常强大的绘图包,基于Wilkinson的《The Grammar of Graphics》一书中提出的图形语法理论而开发,作者是Hadley Wickham。ggplot2的设计思想是将数据集映射到图形属性上,如颜色、形状、大小等,从而可以很方便地绘制出漂亮的图形。其核心理念是将绘图与数据分离,数据相关的绘图与数据无关的绘图分离,是按图层作图,同时它保有命令式作图的调整函数,使其更具灵活性。
- reshape2 R包
reshape2主要功能是在宽格式(wide-format)和长格式(long-format)之间轻松转换数据。宽格式数据通常指的是每个变量单独成一列的数据结构,而长格式数据则是将变量ID整合在同一列中,每行代表一个观察值,每列代表一个变量。
reshape2包主要有两个核心函数:melt()和cast()(或者dcast(),取决于具体的使用情况)。
- melt()函数用于将数据从宽格式转换为长格式。可以想象成我们在处理金属,当熔化金属成液体滴下时,金属会被拉长(long-format)。
- cast()或dcast()函数则用于将长格式数据“重铸”成宽格式数据。如果我们把金属铸成一个模子,它就会变宽(wide-format)。
- circlize R包
circlize是一个用于绘制环形图的R语言包,由德国癌症中心的华人博士Zuguang Gu开发。它并不是生成Circos配置文件的前端封装,而是完全基于R语言风格的统计和图形语法实现的。circlize的设计原理在于其圆形布局由sectors(扇形)和tracks(轨迹)组成,对于数据中的不同分类,它们会作为不同的sectors,而同一个类别的不同维度的观测值,则会作为从圆外向圆内不断堆叠的图形轨迹tracks。
二、 代码实操
废话不多说,小果直接带小伙伴们来实操代码,进行序列分析的全面示例,展示如何使用R语言进行序列比对和可视化!本次介绍的R包操作会占用内存较大,小果建议使用服务器,欢迎小伙伴们联系小果租赁性价比居高的服务器!
- 序列定义:先用Biostrings包定义两个DNA序列s1和s2。
library(Biostrings)
s1<-DNAString(“ACTTCACCAGCTCCCTGGCGGTAAGTTGATCAAAGGAAACGCAAAGTTTTCAAG”)
s2<-DNAString(“GTTTCACTACTTCCTTTCGGGTAAGTAAATATATAAATATATAAAAATATAATTTTCATC”)
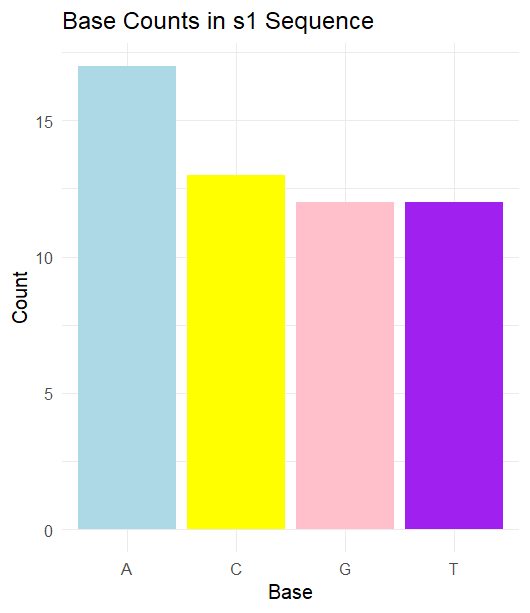
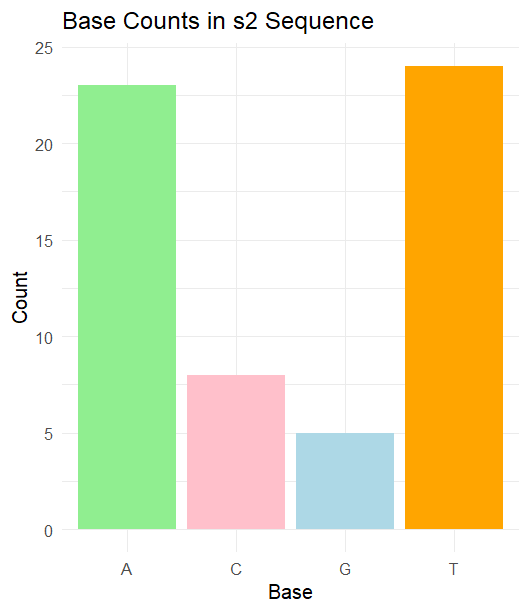
- 碱基计数:计算每个序列中每个碱基(A, C, G, T)的数量,并生成条形图。
base_counts_s1 <- tapply(unlist(strsplit(as.character(s1), “”)), unlist(strsplit(as.character(s1), “”)), length)
base_counts_s2 <- tapply(unlist(strsplit(as.character(s2), “”)), unlist(strsplit(as.character(s2), “”)), length)
base_counts_df1 <- data.frame(
Base = names(base_counts_s1),
Count = base_counts_s1
)
base_counts_df2 <- data.frame(
Base = names(base_counts_s2),
Count = base_counts_s2
)
library(ggplot2)
ggplot(base_counts_df1, aes(x = Base, y = Count)) +
geom_bar(stat = “identity”, fill = c(“lightblue”, “yellow”, “pink”, “purple”)) +
theme_minimal() +
labs(title = “Base Counts in s1 Sequence”, x = “Base”, y = “Count”)
ggplot(base_counts_df2, aes(x = Base, y = Count)) +
geom_bar(stat = “identity”, fill = c(“lightgreen”, “pink”, “lightblue”, “orange”)) +
theme_minimal() +
labs(title = “Base Counts in s2 Sequence”, x = “Base”, y = “Count”)
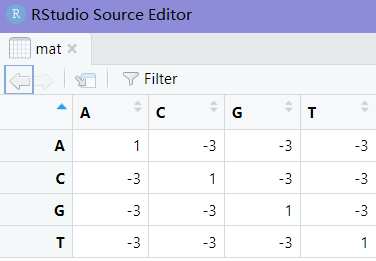
- 全局、局部和重叠序列比对:小果使用pairwiseAlignment函数进行三种不同的序列比对(全局、局部和重叠),并使用了自定义的核苷酸替换矩阵。
mat <- nucleotideSubstitutionMatrix(match = 1, mismatch = -3, baseOnly = TRUE)
globalAlign <-pairwiseAlignment(s1, s2, substitutionMatrix = mat,
gapOpening = 5, gapExtension = 2)
localAlign <-pairwiseAlignment(s1, s2, type = “local”, substitutionMatrix = mat,
gapOpening = 5, gapExtension = 2)
overlapAlign <-pairwiseAlignment(s1, s2, type = “overlap”, substitutionMatrix = mat,
gapOpening = 5, gapExtension = 2)
- 质量评分和模糊矩阵:小果对序列进行带有质量评分和模糊矩阵的比对(在处理测序数据时很有用)
pairwiseAlignment(s1, s2,
patternQuality = SolexaQuality(rep(c(22L, 12L), times = c(36, 18))),
subjectQuality = SolexaQuality(rep(c(22L, 12L), times = c(40, 20))),
scoreOnly = TRUE)
pairwiseAlignment(s1, s2,
patternQuality = SolexaQuality(rep(c(22L, 12L), times = c(36, 18))),
subjectQuality = SolexaQuality(rep(c(22L, 12L), times = c(40, 20))),
type = “local”)
mapping <- diag(4)
dimnames(mapping) <- list(DNA_BASES, DNA_BASES)
mapping[“C”, “T”] <- mapping[“T”, “C”] <- 1
mapping[“G”, “A”] <- mapping[“A”, “G”] <- 1
pairwiseAlignment(s1, s2,
patternQuality = SolexaQuality(rep(c(22L, 12L), times = c(36, 18))),
subjectQuality = SolexaQuality(rep(c(22L, 12L), times = c(40, 20))),
fuzzyMatrix = mapping,
type = “local”)
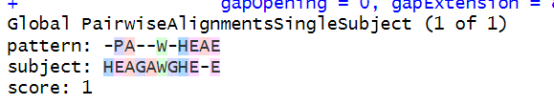
pairwiseAlignment(AAString(“PAWHEAE”), AAString(“HEAGAWGHEE”),
substitutionMatrix = “BLOSUM50”,
gapOpening = 0, gapExtension = 8)
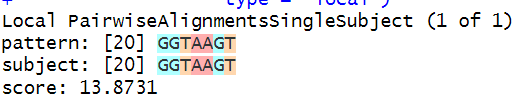
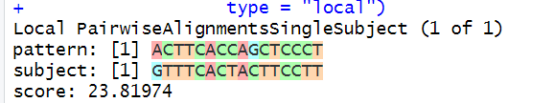
- 序列比对结果打印:打印出全局比对、局部比对和重叠比对的结果。
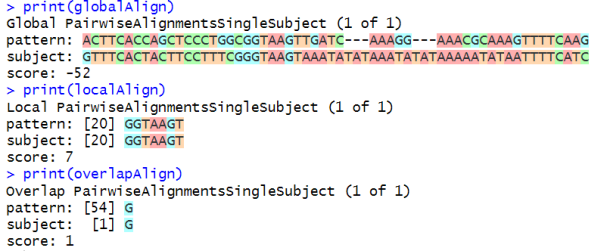
- 比对得分:计算并比较不同比对类型的得分,并将结果存储在scores_df数据框中。
globalScore <- globalAlign@score
localScore <- localAlign@score
overlapScore <- overlapAlign@score
scores_df <- data.frame(
Alignment = c(“Global”, “Local”, “Overlap”),
Score = c(globalScore, localScore, overlapScore)
)
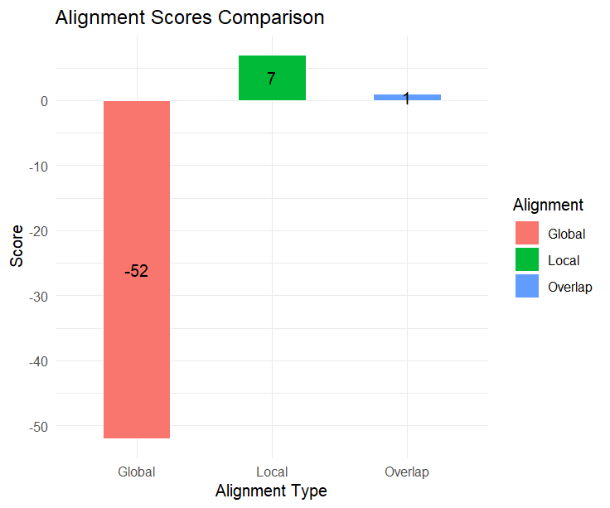
- 得分比较图表:使用ggplot2生成条形图比较不同比对类型的得分。
ggplot(scores_df, aes(x = Alignment, y = Score, fill = Alignment)) +
geom_bar(stat = “summary”, fun = “mean”,color = “white”, width = 0.5) +
geom_text(aes(label = Score), position = position_stack(vjust = 0.5),color=’black’)+
theme_minimal() +
labs(title = “Alignment Scores Comparison”, x = “Alignment Type”, y = “Score”)
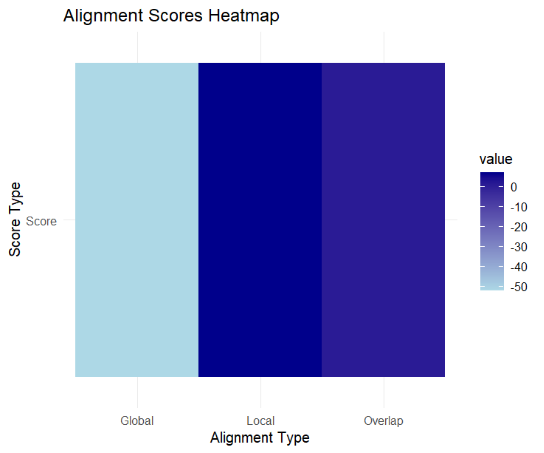
- 得分热图:使用reshape2将scores_df数据框重塑,然后用ggplot2生成得分热图。
library(reshape2)
melted_scores <- melt(scores_df)
ggplot(melted_scores, aes(x = Alignment, y = variable, fill = value)) +
geom_tile() +
theme_minimal() +
scale_fill_gradient(low = “lightblue”, high = “darkblue”) +
labs(title = “Alignment Scores Heatmap”, x = “Alignment Type”, y = “Score Type”)
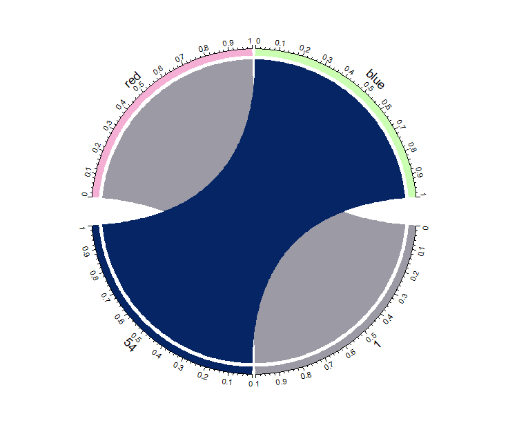
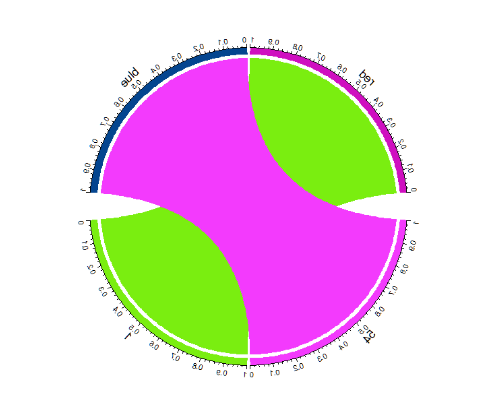
- 安装和加载circlize包绘制和弦图:小果使用chordDiagram函数生成两个序列的和弦图,展示序列之间的相似性和差异性。
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“circlize”)
library(circlize)
genome <- data.frame(breaks = c(1, nchar(as.character(s1))),
cols = c(“red”, “blue”),
labels = c(“Sequence 1”, “Sequence 2”),
stringsAsFactors = FALSE)
chordDiagram(genome, transparency = FALSE)
genome <- data.frame(breaks = c(1, nchar(as.character(s2))),
labels = c(“Sequence 1”, “Sequence 2”),
stringsAsFactors = FALSE)
chordDiagram(genome, transparency = FALSE)
三、 文章小结
本期小果和小伙伴们一起学习了R语言进行序列分析的全面示例,学习了如何使用R语言进行序列比对,掌握了数据处理的关键技巧。此外,我们还学习了如何将分析结果以直观的方式呈现出来,实现了数据的可视化。这不仅加深了我们对生物信息学知识的理解,也锻炼了我们的R语言实践能力。希望本期内容对小伙伴们有帮助~让我们继续探索生信的无限可能,共同开启更多科研新篇章!最后如果各位小伙伴们觉得自己运行代码太麻烦,欢迎用我们的云生信小工具,只要输入合适的数据就可以直接出想要的图哦,附云生信链接(http://www.biocloudservice.com/home.html)。