Hello! 今天我很高兴地向大家介绍一个令人振奋的R包——RNAseqStat2。这款工具简直是生物信息学分析的利器,只需一行命令,便能轻松应对RNA-seq的下游分析挑战。接下来,小果将带你们进入RNAseqStat2的实战应用,分分钟教会你们如何得到漂亮的结果图!作为在生物信息学领域摸爬滚打多年的我,非常乐意与大家分享我的经验和知识。若您在进行生物信息学分析时遇到难题,随时欢迎向我寻求帮助。
如何使用一行代码便捷地进行差异分析和富集分析呢?别急,今天小果就用一段代码来示范,手把手教你们如何驾驭RNAseqStat2,让你们在RNA-seq数据分析的征途上,轻松愉快地收获那些令人惊艳的可视化成果!
在我们深入探索R包的无限可能时,小果注意到,某些强大的R包在运行时可能会对您的计算机内存提出较高的要求。小果在这里为您提供了一系列性价比极高的服务器租赁选项,旨在满足您从轻量级到重量级的数据分析需求。
- 一:安装以及加载R包
library(devtools)
devtools::install_github(“xiayh17/RNAseqStat2”)
- 二:模式生物,一步完成分析
模式生物只需要按照示例数据整理文件即可,下载对应的OrgDB包,一行命令即可出图,非常的简单。如果是Human, Mouse,Rat 之一,整体分析非常简单,短短几行即可完成全部分析,以airway的数据为例(R中的airway数据集是一个RNA测序数据集,它包含了四个人类气道平滑肌细胞系(airway smooth muscle cell lines)在接受地塞米松(dexamethasone,一种糖皮质激素)处理后的基因表达数据。
library(RNAseqStat2)
library(airway)
data(“airway”)
row_counts <- as.data.frame(assay(airway))
group_list <- as.character(colData(airway)$dex)
data_i <- Create_DEGContainer(species = “Human”,
dataType = “Counts”,
idType = “ENSEMBL”,
expMatrix = row_counts,
groupInfo = group_list,
caseGroup = “trt”)
# 一步运行
data_o <- runALL(object = data_i,dir = “output_test”)
在这里小果提醒大家,如果是其他物种,需要根据科学问题而设置参数,下载对应的OrgDB包。
data_i <- Create_DEGContainer(species = “pig”,
dataType = “Counts”,
idType = “SYMBOL”,
expMatrix = row_counts,
groupInfo = group_list,
caseGroup = “NBW”,
OrgDb=”org.Ss.eg.db”,
organism=”ssc”,
msi_species=”Sus scrofa”)
- 三:非模式物种构建对象
小众的非模式生物甚至都没有合适的Org.Db数据库。这个时候就需要自己准备一些文件,包括注释文件等。小果这边来详细讲解一下。
首先需要准备5个初始文件,包括原始表达量矩阵和注释文件。对于非模式植物,获取GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)注释文件的方法通常涉及以下几个方法:一、使用EGGNOG-Mapper进行注释:对于非模式物种,可以使用EGGNOG-Mapper来快速进行GO或KEGG注释。注释结果文件中将包含序列映射上的GO号或KO号。二、可以将蛋白文件上传到KEGG注释的网站(https://www.genome.jp/tools/kofamkoala)中进行注释。初始文件如下:
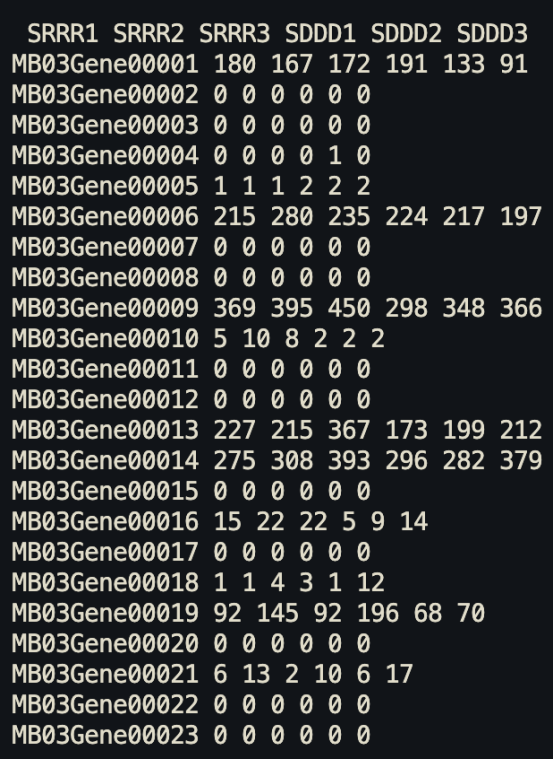
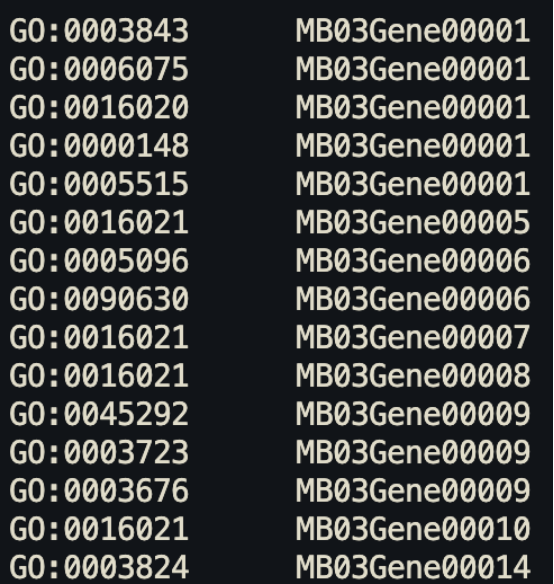
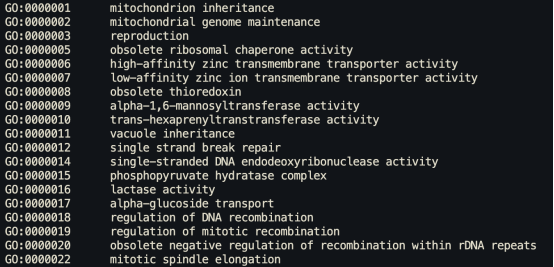
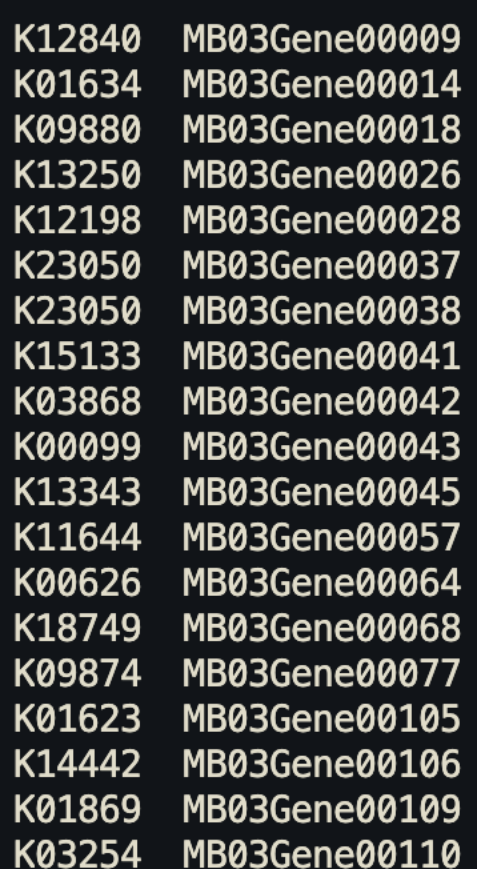
准备好文件即可开始构建对象
# 加载RNAseqStat2包,用于RNA-seq数据分析
library(RNAseqStat2)
# 加载dplyr包,用于数据操作和处理
library(dplyr)
# 读取行计数数据,这里假设数据是以空格分隔的,并且第一行包含列名,行名为数据的第一列
# 请根据实际文件格式调整sep参数(例如,如果是逗号分隔,使用”,”)
row_counts <- read.delim(“SRD.txt”, header=TRUE, sep = ” ” , row.names=1)
# 定义分组列表,每个元素对应一个样本的分组
# 这里的分组名称是示例,应根据实际样本分组进行修改
group_list <- c(“SRRR”,”SRRR”,”SRRR”,”SDDD”,”SDDD”,”SDDD”)
# 读取基因的GO注释文件,这里假设文件是以制表符分隔的,没有列名,不检查基因名的有效性
# 请根据实际文件格式调整sep参数和header参数
GO_anno <- read.csv(“gene_go_annotation_from_ipr_nr.tab”, sep = “\t”, header = FALSE, check.names = FALSE)
# 读取GO术语的描述文件,同样假设没有列名,不检查术语名的有效性
GO_description <- read.csv(“go_term.list”, sep = “\t”, header = FALSE, check.names = FALSE)
# 读取基因的KEGG注释文件,假设文件格式与GO注释文件相同
KEGG_anno <- read.csv(“kegg_gene.txt”, sep = “\t”, header = FALSE, check.names = FALSE)
# 读取KEGG路径的描述文件,假设文件格式与GO描述文件相同
KEGG_description <- read.csv(“kegg_description.txt”, sep = “\t”, check.names = FALSE, header = FALSE)
#构建对象
data_i <- Create_DEGContainer(expMatrix = row_counts,
groupInfo = group_list,
caseGroup = “SDDD”,
species = “unknow”,
GOTERM2GENE = GO_anno,
GOTERM2NAME = GO_description,
KEGGTERM2GENE = KEGG_anno,
KEGGTERM2NAME = KEGG_description)
#一步法完成数据分析
data_o <- runALL(object = data_i,dir = “output”)
#这里小果给大家介绍一下函数包含的参数:
expMatrix 处理好的表达矩阵
species 物种名 比如 Human Mouse Rat 或者其他
groupInfo 分组信息 每个样本对应哪个分组
caseGroup 实验组 哪个分组是实验组
GOTERM2GENE、GOTERM2NAME、KEGGTERM2GENE、KEGGTERM2NAME为注释文件信息
MSigDB(Molecular Signatures Database)是一个包含成千上万个注释基因集的数据库,分为人类和鼠类基因集,并且可以通过其官方网站进行访问和使用。我们的物种为非模式生物,这步会出现提醒,可以忽略。
object为构建的对象,dir设置结果文件的保存路径,结果包括样本间的PCA图、热图、分布图、火山图、top差异基因的表达热图、多种方法的Venn图等。

l 四:结果解读
构建好对象后仅需一步即可完成全部的下游分析!接下来小果带大家解读一下结果哦~
1-run_check
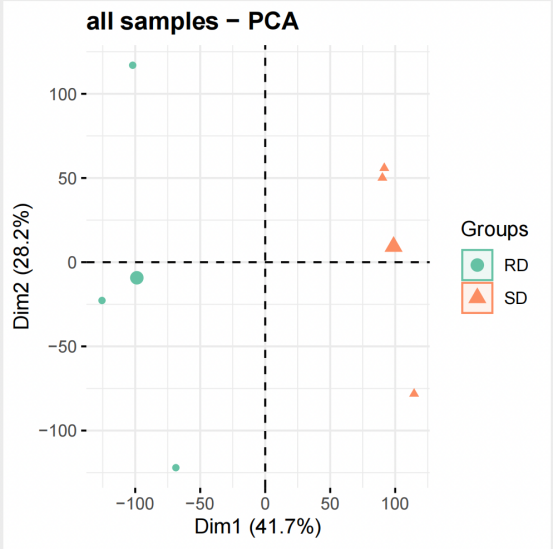
PCA图
横纵坐标:两个维度
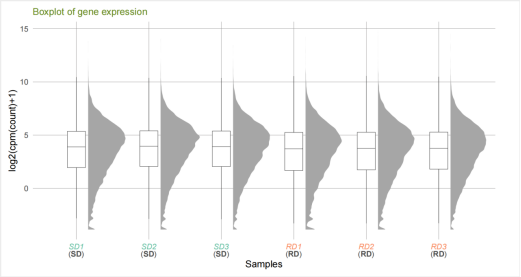
boxplot图
横坐标:样品名
纵坐标:log2(cpm(count)+1)
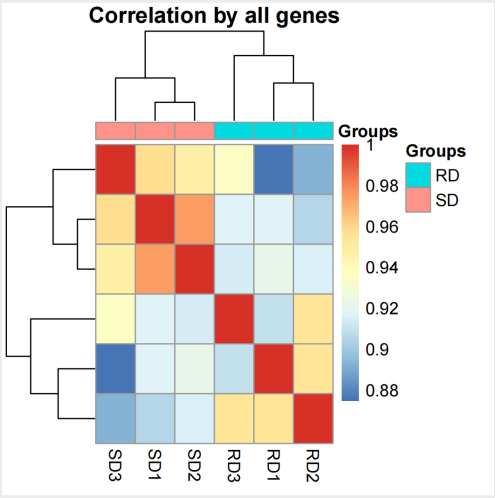
相关性图
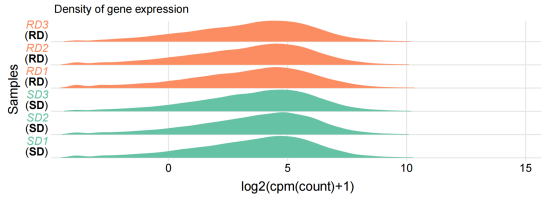
分布图
boxplot图的另一个视角,转换了x,y轴
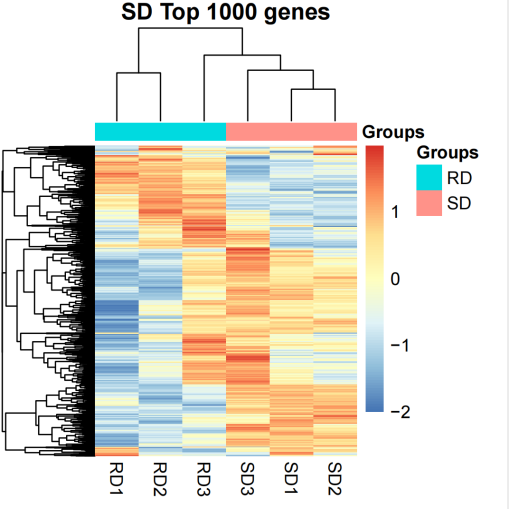
热图
横坐标:样本
纵坐标:基因
前1000个基因绘制热图
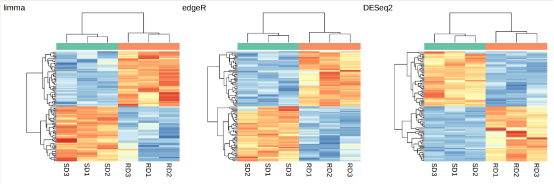
2-runDEG
热图
三种方案前500个基因绘制的热图
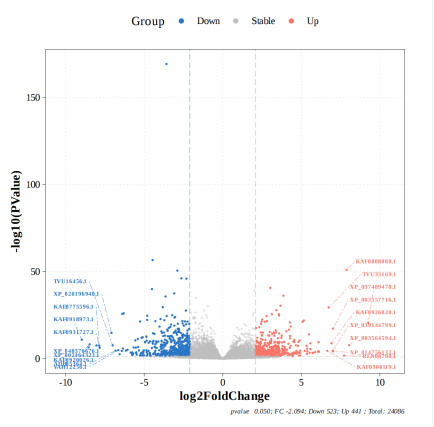
火山图
横坐标:log2(Fold Change)
纵坐标:-log10(p-value)
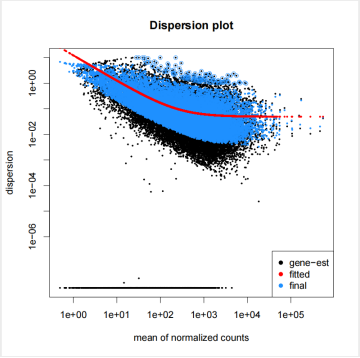
离散图
横坐标:标准count数的平均值
纵坐标:离散度
质控RAWvsNORM图
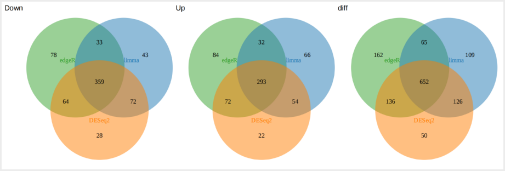
维恩图
三个图分别为下调基因、上调基因和差异基因
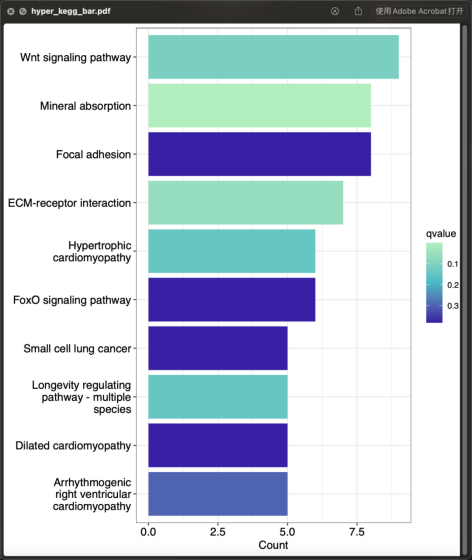
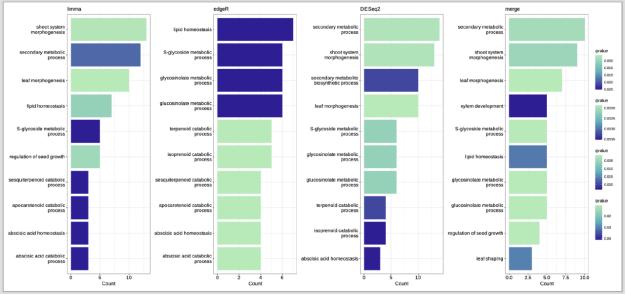
3-runHyper
富集分析结果的柱状图

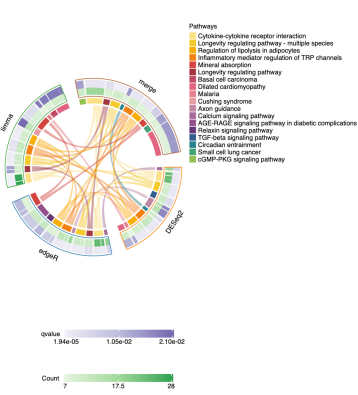
圈图
外围紫色代表q值,往内绿色代表count数,再往内彩色代表不同的通路,内部相同的通路用线连接。
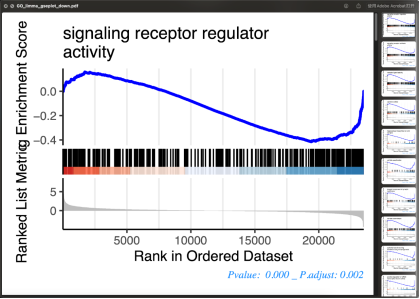
4-runGSEA
标题:表示不同通路
第一部分:
折线图:离垂直距离x=0轴最远的峰值便是基因集的ES值,峰出现在排序基因集的前端(ES值大于0)则说明通路上调,出现在后端(ES值小于0)则说明通路下调。
第二部分:
中间从蓝色到红色的过渡“带”表示基因从上调到下调排列(排序可以按照fold change,也可以是p-value)。黑色像条形码的竖线表示该位置的基因属于某个指定通路。绿色有波动的曲线表示富集分数,从0开始计算,属于基因通路增加,不属于则减少。最后看下黑色的条形码是不是富集在一端。
第三部分:
表示每个基因对应的信噪比(Signal2noise)。
排序后所有基因rank值(由log2FoldChang值计算得出)的分布,以灰色面积图显展示。
以上内容便是对RNAseqStat2包的详尽介绍及其基本使用方法的演示,怎么样,你学会了嘛!?如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便,云平台网址为:(http://www.biocloudservice.com/home.html)
小果今天的分享就暂告一段落,非常期待与你一起探讨学习更多生物信息学的知识!如果你有任何疑问或建议,随时欢迎与小果交流。我们下期再见,期待你的再次光临!