
嗨,我是小师妹,很高兴能够和大家一起来学习生物信息学。前几天小师妹给大家带来了多序列比对工具mafft的分享。今天,小师妹就继续带来系统发育分析一站式的分享!今天给大家介绍的是一个非常强大的工具——IQ-TREE2。本次介绍的软件操作占用内存比较大,建议使用服务器,欢迎联系小师妹租赁性价比居高的服务器~

做遗传进化的朋友肯定对进化树不会陌生,现在文献市场上做系统发育的这么多,要问进化树构建哪家强,公众号关注R语言学徒找答案。IQ-TREE2专为系统发育推断而设计,提供了快速准确的树重建功能。这款软件以其高效的算法和用户友好的界面,使得即使是处理大规模数据集也变得轻松快捷。进化树可难不倒生信高手小师妹,要是同学们有自己做不了的生信分析,欢迎随时联系我!!!。
自2013年推出以来,iqtree以其快速精准的建树和全面的功能在竞争激烈的系统发育树(phylogenetics)领域快速蹿红,并广受好评,目前砍下谷歌学术8748次引用。
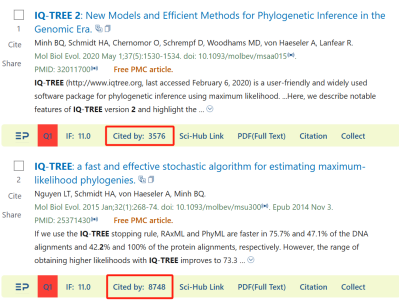
图1 工具引用情况
IQ-TREE2的核心特点包括:
1.高效的树重建:采用快速有效的随机算法,通过最大似然推断系统发育树,计算时间上优于RAxML和PhyML。
2.ModelFinder:这是一个快速准确的模型选择工具,支持自由速率变化模型,并且比jModelTest和ProtTest快10到100倍。它还能找到最合适的分区方案。
3.UFBoot:提供近似无偏的分支支持值,运行速度比非参数引导程序快100倍,比RAxML快速引导程序快10到40倍。
4.AliSim:AliSim(比对模拟器)允许在比 Seq-Gen 和 INDELible 更真实的模型下快速模拟多序列比对。

图2 IQTREE工具的亮点
一、下载和安装
$ wget -c https://github.com/iqtree/iqtree2/releases/download/v2.1.3/iqtree-2.1.3-Linux.tar.gz
$ tar -zxvf iqtree-2.1.3-Linux.tar.gz
$ echo ‘export PATH=”~/iqtree-2.1.3-Linux/bin:$PATH”‘>>~/.bashrc
$ source .bashrc
解压后iqtree执行文件就在bin/目录下,进入iqtree-2.1.3-Linux目录后还有一个示例文件example.phy。
二、常用命令
1.选择最佳模型
IQTREE2内置了上百种模型,作为小白的我们应该如何选择呢,不用担心,我们可以利用ModelFinder功能进行最佳模型选择,参数-m设置MF自动选择最佳模型但不建树;参数-m设置MFP自动检测最佳模型并建树。
$ iqtree2 -s example.phy -m MF
图3 ModelFinder结果文件
Iqtree会测试多达546个蛋白模型并给出最佳模型,打开后缀名为model.gz,结果如图4所示:
$ zcat example.phy.model.gz | less
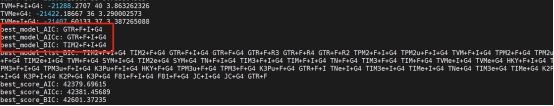
图4 ModelFinder结果示意图
我们找到红色方框标注部分,就是ModelFinder的最终结果。依据AIC准则,最佳的替换模型应该为GTR+F+I+G4;依据BIC准则,最佳的替换模型为TIM2+F+I+G4。我们可以依据个人的研究需求以及下一步的工作选择合适的准则,如果需要进行贝叶斯推断(构建MCMC系统发育树),那我们应该采用贝叶斯(BIC)准则。
此外还有:
example.phy.iqtree:主要报告文件,我们可以查看此文件以查看计算结果。它还包含最终树的文本表示(见下文)。
example.phy.treefile:NEWICK格式的ML树,可以通过任何支持的树查看器程序(如FigTree或iTOL)进行可视化。
example.phy.log:整个运行的日志文件。
$ less example.phy.iqtree
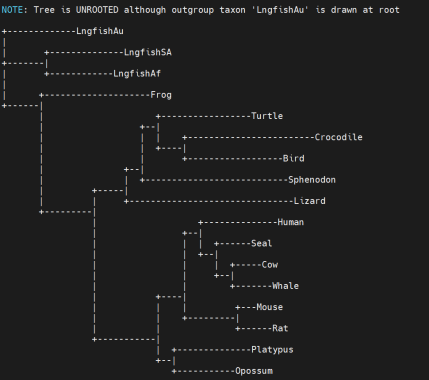
图5 example.phy.iqtree主要报告文件
2.构建系统发育树
得到了最佳模型之后,就可以进行系统发育树的构建了,这里选择的是赤池信息(AIC)准则选择的最佳模型。
$ iqtree2 -s example.phy -m GTR+F+I+G4
3.其他命令
直接选择最佳的模型进行树的构建,不进行bootstrap:
$ iqtree.exe -s example.phy -m MFP
进行最佳模型构建树,并且进行bootstrap:
$ iqtree.exe -s example.phy -m MFP -b 1000 -nt AUTO
进行GTR+F+I+G4模型构建系统发育树:
$ iqtree.exe -s example.phy -m GTR+F+I+G4 -b 1000 -nt AUTO
重抽样加速,结果无差异,但是耗时相对少一些:
$ iqtree.exe -s example.phy -m GTR+F+R3 -b 1000 -bnni -nt AUTO -redo
使用4个 CPU 核来加速计算:
$ iqtree -s example.phy -T 4
基于IQTree的UFBoot2进行打分,这个打分并不是普通的Bootstrap自展值,而是一种近似值,速度会快很多,但要在95%以上的UFBoot才可信。如果想要改成普通的Bootstrap值,则改成-b 100,我们平时查看进化树可以不设置自展值,但是如果要发表文献,要记得设置1000以上的自展值,编辑才会认同哦~
$ iqtree -s example.phy -m MFP -B 1000
–symtest:目前一般的建树方法,都是基于所谓的SRH原则,即stationary, reversible, and homogeneous,对于SRH原则的违背在文献中十分普遍,建出来的树是否真的可靠就要打上问号了。你的dataset是否存在对该原则的违背呢?不妨用–symtest检测一下。
$ iqtree -s example.phy -symtest
2.0.3版本中增加了LSD2法对进化时间的推算。不过目前有两个不足之处,一是给出的结果属于点估计,二是当不同序列或物种间进化速率差异较大时,可能不太灵光。
三、小结
既然说到建树基于多序列比对,就不得不介绍一下整体流程:
①挑选合适的基因生成fasta文件(DNA/AA)
②多序列比对 (Mafft/ ClustalW)
③去掉比对中的gap (TrimAl)
④转换格式,生成的FASTA, NEXUS, CLUSTALW格式的文件均
⑤建树 (IQ-TREE)
⑥进化树美化(figtree、itol、mega)
今天小师妹给大家介绍的是第五步,其中树文件为后缀是treefile,导入mega或itol即可进行可视化,结果如下。
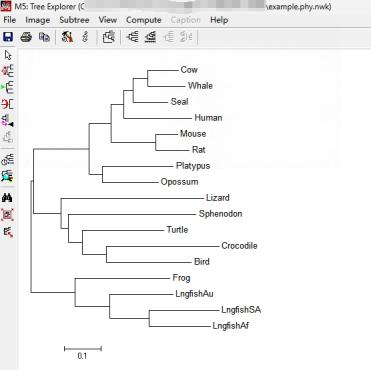
图6 Mega软件可视化进化树
把treefile(nwk文件)导入到树查看程序就可以进行进化树的美化啦,我们可以用iTol、figtree等等,以上代码就是对IQTree2的介绍与基本使用方法,学会这些方法后,你可以学习如何使用IQTree2进行系统发育树的绘制,包括选择最佳的替换模型、设置Bootstrap值等等。当然,大家也可以根据自己的需求和研究目标修改和扩展这些代码哦!!怎么样,你学会了嘛!?
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!