科研小伙伴们,面对堆积如山的WGBS数据,是否感到无从下手?别担心,有了bsseq包,我们可以轻松驾驭BSmooth算法,让复杂的数据分析变得简单。
全基因组甲基化测序(Whole Genome Bisulfite Sequencing, WGBS)是一种用于检测DNA甲基化状态的高通量测序技术。DNA甲基化是一种重要的表观遗传修饰,它在调控基因表达、胚胎发育、细胞分化以及许多疾病的发生发展中扮演着关键角色。通过WGBS技术,研究人员可以在整个基因组范围内精确地定位甲基化位点,测量特定基因组区域的甲基化水平,这对于理解基因表达调控、疾病发展乃至生物进化都至关重要。
而bsseq包就是一个专门用于WGBS数据分析的工具,它能帮助我们从海量数据中精准识别差异甲基化区域(DMRs)。该算法自Hansen等人的研究中首次亮相以来,已经成为我们分析WGBS数据的得力助手。
今天,小师妹将带领大家从基础的数据准备开始,逐步深入到高级分析技巧,无论你是生物信息学的新手还是资深研究者,bsseq包都将是你的得力助手。注意哦,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小师妹租赁性价比高的服务器~
准备好了吗?让我们携手踏上这场基因组甲基化分析的奇妙之旅,一起探索基因组的奥秘,揭开生命科学中那些隐藏的秘密。旅途中遇到任何难题,记得找小师妹哦,我随时准备着帮你答疑解惑!
一、 加载数据
首先,让我们开始安装bsseq包和另外需要用到的包:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“bsseq”)
接下来,我们需要加载`bsseq`和`bsseqData`库,然后加载并更新`BS.cancer.ex`数据集,它包含了未经平滑处理的“原始”汇总比对数据。
library(bsseq)
library(bsseqData)
# 加载BS.cancer.ex数据集
data(BS.cancer.ex)
# BS.cancer.ex <- updateObject(BS.cancer.ex)
BS.cancer.ex

`BS.cancer.ex`对象包含了958,541个甲基化位点和6个样本,这些数据尚未进行平滑处理,所有分析都在内存中进行。通过查看`pData(BS.cancer.ex)`,我们可以了解到这些样本的类型和配对信息。每个样本都被标记为癌症(cancer)或正常(normal),并分配了一个配对编号(pair1, pair2, pair3)。
# 查看样本数据框架
pData(BS.cancer.ex)

输出显示了一个包含6行2列的数据框架,其中包含了样本类型和配对信息。例如,C1、C2和C3代表癌症样本,而N1、N2和N3代表正常样本,它们分别与pair1、pair2和pair3配对。
二、 数据平滑处理
在分析的第一步中,我们需要对数据进行平滑处理以减少噪声并提高数据的可读性。
BS.cancer.ex.fit <- BSmooth(
BSseq = BS.cancer.ex,
BPPARAM = MulticoreParam(workers = 1),
verbose = TRUE)
在这里,我们先不执行这段代码哦~ 如果你的计算机有6个核心可用,可以将`mc.cores`设置为6,但是Windows系统上不支持将`mc.cores`设置为大于1的值。
为了方便使用,`bsseqData`已经包含了这个命令的结果:
data(BS.cancer.ex.fit)
BS.cancer.ex.fit <- updateObject(BS.cancer.ex.fit)

输出显示了一个`BSseq`类型的对象,包含了958,541个甲基化位点和6个样本,已经使用BSmooth算法进行了平滑处理。
并行化处理
这个步骤使用了并行化处理,每个样本都在单独的核心上运行,使用了`parallel`包中的`mclapply()`函数。这种并行化处理内置于`bsseq`中,需要一台拥有6个核心和足够RAM的计算机。平滑步骤在每个样本上完全独立进行,因此如果有很多样本(或其他情况),也可以手动分割计算。
CpG位点的覆盖度
在全基因组甲基化测序中,CpG位点的覆盖度是一个关键指标。
`BS.cancer.ex`对象包含了人类21号和22号染色体上所有注释的CpG位点。因为我们有多个样本,我们可以将基因组大致分为三类:所有样本都有数据的CpG位点、所有样本都没有数据的CpG位点,以及部分样本有数据的CpG位点。检查当前对象,我们可以得到:
# 两个染色体上CpG位点的平均覆盖度
round(colMeans(getCoverage(BS.cancer.ex)), 1)

# CpG位点的数量
length(BS.cancer.ex)
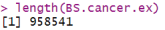
# 所有6个样本中至少被1个读取覆盖的CpG位点数量
sum(rowSums(getCoverage(BS.cancer.ex) >= 1) == 6)
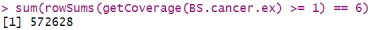
# 所有样本中覆盖度为0的CpG位点数量
sum(rowSums(getCoverage(BS.cancer.ex)) == 0)

由此我们可以知道覆盖度大致为4倍,因此我们预期会有很多覆盖度为0的CpG位点。但是,不是说每个CpG位点在所有6个样本里都会覆盖度为零,这事儿没那么绝对。
为了更好地理解这一点,我们可以假设全基因组的覆盖度遵循参数为4的泊松分布。在这种情况下,我们可以计算至少有一个样本覆盖度为零的CpG位点的预期比例。
# 计算泊松分布的概率
logp <- ppois(0, lambda = 4, lower.tail = FALSE, log.p = TRUE)# 计算至少有一个样本覆盖度为零的CpG位点的比例
round(1 – exp(6 * logp), 3)

这段代码的输出结果表明,我们预期大约有10.5%的CpG位点至少在一个样本中的覆盖度为零。这个计算有助于我们理解数据中的零覆盖现象,并在后续分析中对这些CpG位点进行适当的处理。
我们的数据里,有好些CpG位点在六个样本里都没啥信息,差不多有13万个吧。这可能是因为这些位点太傲娇了,不容易被映射到基因组上。我们现在用的还是50bp的单端测序,要是换成100bp的配对末端测序,能映射的基因组区域会多不少呢。对于那些在所有样本中覆盖度都为零的CpG位点,我们处理起来还算简单,但得出结论时得悠着点。
还有啊,有些CpG位点在部分样本中默默无闻,在部分样本中覆盖度为零,但不是全部。这些位点就棘手了,因为我们的覆盖度本来就不高,偶尔一个样本没覆盖到也是正常的,要是直接排除这些位点,可能就太武断了。
平滑处理的技巧
接下来说说平滑处理。我们得对每个样本单独做平滑,只用那些覆盖度非零的位点。这样就能估计出全基因组的甲基化情况,然后给BSseq对象里的每个CpG位点都估个甲基化值。这招在比较不同样本的同一个CpG位点时特给力,尤其是那些偶尔覆盖度为零的样本。不过,如果基因组某些区域附近压根就没覆盖到CpG位点,那平滑后的甲基化轮廓就没多大意义了。我们可以在平滑后把这些位点剔除掉。
手动分割平滑计算
以下是一个手动分割平滑计算示例,在这里我们先只使用样本1和2:
# 分割数据
BS1 <- BS.cancer.ex[, 1]
save(BS1, file = “BS1.rda”)
BS2 <- BS.cancer.ex[, 2]
save(BS1, file = “BS1.rda”)
# 在每个节点上执行以下操作
# 节点1
load(“BS1.rda”)
BS1.fit <- BSmooth(BS1)
save(BS1.fit)
save(BS1.fit, file = “BS1.fit.rda”)
# 节点2
load(“BS2.rda”)
BS2.fit <- BSmooth(BS2)
save(BS2.fit, file = “BS2.fit.rda”)
# 合并(在新的R会话中)
load(“BS1.fit.rda”)
load(“BS2.fit.rda”)
BS.fit <- combine(BS1.fit, BS2.fit)
注意哦,这仍需要有一个节点有足够的RAM来容纳所有样本。
三、计算t统计量
在计算t统计量之前,我们得先清理一下数据。那些覆盖度很低或者根本没有覆盖的CpG位点,我们就不要了。要是不这么做,你可能会在覆盖度很低的基因组区域找到很多差异甲基化区域(DMRs),这些很可能是假阳性。具体要去掉哪些CpG位点,这得看个人喜好,但在这个分析中,我们只保留至少有两个癌症样本和至少有两个正常样本覆盖度至少为2倍的CpG位点。为了看起来更清楚,我们把覆盖度数据存在一个单独的矩阵里。
BS.cov <- getCoverage(BS.cancer.ex.fit)
keepLoci.ex <- which(rowSums(BS.cov[, BS.cancer.ex$Type == “cancer”] >= 2) >= 2 &
rowSums(BS.cov[, BS.cancer.ex$Type == “normal”] >= 2) >= 2)
length(keepLoci.ex)

BS.cancer.ex.fit <- BS.cancer.ex.fit[keepLoci.ex,]
现在,我们已经可以开始计算t统计量了。
BS.cancer.ex.tstat <- BSmooth.tstat(BS.cancer.ex.fit,
group1 = c(“C1”, “C2”, “C3”),
group2 = c(“N1”, “N2”, “N3”),
estimate.var = “group2”,
local.correct = TRUE,
verbose = TRUE)
BS.cancer.ex.tstat
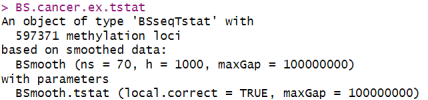
t统计量的计算过程
`BSmooth.tstat()`函数的参数挺简单的。`group1`和`group2`包含了两组被比较的样本名称(总是`group1`减去`group2`),也可以用索引代替样本名称。`estimate.var`参数描述了用哪些样本来估计变异性。因为这个是癌症数据集,癌症样本的变异性比正常样本高,所以我们只用正常样本来估计变异性。`estimate.var`的其他选择有`same`(假设每组的变异性相同)和`paired`(做一个配对t检验)。`local.correct`参数描述了我们是否应该使用大规模(低频)均值校正。这在癌症研究中尤其重要,因为我们发现癌症和正常组织之间存在很多大规模的甲基化差异。
我们可以通过以下代码查看t统计量的边际分布:
plot(BS.cancer.ex.tstat)
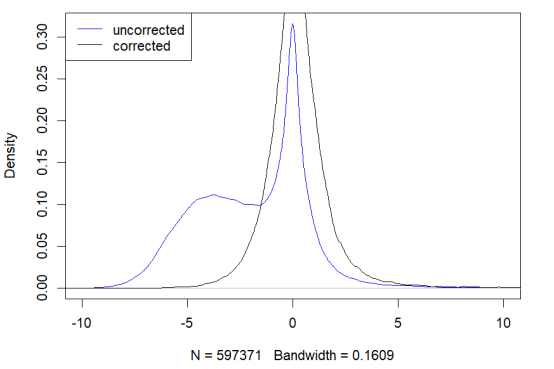
在未校正的t统计量的边际分布中,可以清楚地看到低甲基化“块”。
即使在没有观察到这些大规模甲基化差异的比较中,对t统计量进行局部校正通常也会改善它们的边际分布(“改善”是指使它们更加对称)。
校正t统计量的意义
局部校正t统计量的意义在于,它能帮助我们得到更加准确的甲基化差异区域。通过这种方法,我们可以减少由于偶然因素或实验误差导致的假阳性结果,从而更有信心地指出真正的生物学差异。这种校正方法在处理大规模甲基化数据时尤为重要,因为它可以帮助我们识别出那些在癌症和正常组织之间存在显著差异的区域。
四、寻找差异甲基化区域(DMRs)
嘿嘿,说到这儿,小师妹得提一下,等我们把t统计量一算出来,就能划拉个标准线,也就是阈值,来挑出那些甲基化程度不一样的地方,专业点说就是差异甲基化区域,简称DMRs。我们这次划的线是4.6,这个数字可不是拍脑袋定的,是好好瞅了瞅整个基因组的t统计量的分布情况,挑了个合适的分位数值。这样一来,我们就能把那些甲基化程度变化大的区域给标记出来了。
是不是挺有意思的?我们就这么一步步地,把基因组的秘密给揭开啦!
dmrs0 <- dmrFinder(BS.cancer.ex.tstat, cutoff = c(-4.6, 4.6))
## [dmrFinder] creating dmr data.frame
dmrs <- subset(dmrs0, n >= 3 & abs(meanDiff) >= 0.1)
nrow(dmrs)

这段代码的输出告诉我们,我们找到了373个DMRs。我们进一步筛选出那些至少包含3个CpG位点,并且在正常和癌症样本之间的甲基化平均差异至少为0.1的DMRs。
head(dmrs, n = 3)

接下来,小师妹来给你们说说我们怎么挑出那些特别有意思的DMRs。我们先看输出,它会告诉我们前三个DMRs的详细信息,比如它们在基因组的哪个位置,牵涉到多少个CpG位点,区域有多大,还有各种统计数据,比如平均差异、两组样本的平均甲基化水平之类的。这些数据可帮我们大忙了,能让我们清楚地看到哪些区域在正常和癌症样本之间的甲基化程度变化最大。
挑DMRs的时候,我们得定个规矩,比如说,我们就不要那些CpG位点少于3个,或者甲基化差异太小(小于0.1)的区域。虽然这个规矩的具体数字可以商量,但有个标准总是好的。
`dmrFinder()`函数还有个挺酷的功能,就是可以设置`qcutoff`参数,用分位数来定阈值。比如,我们可以设`qcutoff = c(0.01, 0.99)`,这样就能挑出那些甲基化差异特别大的区域。还有个`maxGap`参数,如果两个CpG位点之间的距离超过了这个值,那它们就不会被算作一个DMR。
说到排序,我们可以根据`areaStat`这个指标来,它其实就是每个CpG位点的t统计量加在一起。这个指标有点像是DMR的“面积”,但它是根据CpG位点的数量来加权的,而不是看它们在基因组里占多长。虽然这个方法已经不错了,但我们还是希望能有更好的办法。
总之呢,用这些方法,我们就能更精确地找到那些在癌症和正常组织之间甲基化水平有明显差异的区域,这对于搞生物研究或者想搞点临床应用的师兄师姐们来说,可是超级有用的信息哦!
五、绘制DMRs图
说到查看差异甲基化区域,也就是DMRs,这可是咱们研究中的重要一步呢。为了让这些区域在图上更显眼,咱们可以在pData里加上一些特别的标记,比如颜色、线型和线宽。
就像这样,咱们先设置好pData:
pData <- pData(BS.cancer.ex.fit)
pData$col <- rep(c(“red”, “blue”), each = 3)
pData(BS.cancer.ex.fit) <- pData
然后,咱们就可以开始画图啦,比如画第一个DMR:
plotRegion(BS.cancer.ex.fit, dmrs[1,], extend = 5000, addRegions = dmrs)
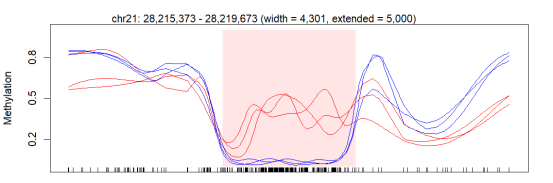
这里的extend就是说,咱们要在图的两边各扩展多少碱基对。而addRegions就是告诉咱们,还有哪些区域需要特别标注一下。
当然了,咱们通常不会只画一个DMR,而是会一次性画上好几百个,然后存到一个PDF文件里,用别的工具慢慢看。这时候,plotManyRegions()函数就派上用场了,它比一个一个地用plotRegion()画要快得多。举个例子:
pdf(file = “dmrs_top200.pdf”, width = 10, height = 5)
plotManyRegions(BS.cancer.ex.fit, dmrs[1:200,], extend = 5000,
addRegions = dmrs)
dev.off()
这段代码就会帮咱们把前200个DMRs都画出来,保存在pdf中啦,这样能让我们在后续的分析或报告中快速回顾和展示我们的发现。这种可视化的方法,在向别人解释复杂的甲基化数据时,特别有用哦。
好啦,咱们今天的甲基化数据分析小课堂就到这里了。小师妹带着大家一起从数据的预处理,到平滑处理,再到找出差异甲基化区域(DMRs),最后还一起看了怎么把这些区域画出来。希望你们都能跟上节奏,对这些复杂的数据分析有了更深的理解。
记得哦,数据分析就像是解谜游戏,每一步都很重要。咱们用到的这些工具和方法,就像是解谜时的小道具,用得好了,就能帮咱们更快地找到答案。所以,大家在实际操作的时候,一定要细心,多尝试,多思考。
如果你们在分析的过程中遇到什么难题,或者有什么新发现,随时欢迎来找小师妹讨论哦。我们一起学习,一起进步。毕竟,科学的道路上,我们从不孤单!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~