嘿,科研小伙伴们,小师妹我又来啦!今天,我要给大家带来一个超级实用的生物信息学工具——Splatter包。在这个单细胞RNA测序数据爆炸的时代,你是不是也在为如何获取和处理这些复杂的数据而头疼?别担心,有了Splatter包,我们可以轻松模拟单细胞RNA测序数据,让数据分析变得更加简单直观。
Splatter是一个R包,专门用于简单模拟单细胞RNA测序数据。这个包的功能非常强大,它不仅可以帮助我们理解数据的特性,还能让我们在实际研究中更加得心应手。通过这个包,我们可以模拟出与真实数据极为相似的单细胞数据集,这对于验证分析方法、测试新的算法或进行教学演示都是非常有用的。
在这个教程中,小师妹将带你一步步了解Splatter包的安装、快速入门指南,以及如何进行参数估计和数据模拟。注意哦,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小师妹租赁性价比高的服务器~
准备好了吗?让我们一起开启这场单细胞数据分析的奇妙之旅吧!如果在旅途中遇到任何问题,记得找小师妹帮忙哦!
首先,让我们开始安装multistateQTL包和另外需要用到的包:
if (!require(“BiocManager”, quietly=TRUE))
install.packages(“BiocManager”)
BiocManager::install(“splatter”)
BiocManager::install(“scater”)
安装好了之后,记得加载这些必要的库:
lsuppressPackageStartupMessages({
library(splatter)
library(scater)
})
一、快速上手 Splatter包
如果你已经有了一个类似于你想要模拟的计数数据集,那么只需要两步,我们就能创建一个模拟的数据集。下面是如何用`scater`包生成一个模拟数据集的例子:
# 创建模拟数据
set.seed(1)
sce <- mockSCE()
# 从模拟数据估计参数
params <- splatEstimate(sce)

这一步,Splatter会提醒我们,它发现库大小是正态分布而不是对数正态分布。这个提示很有用,因为它告诉我们数据的分布特性,我们可以根据这个信息调整模拟参数。
接下来,我们用估计出的参数来模拟新的数据集:
# 使用估计的参数模拟数据
sim <- splatSimulate(params)
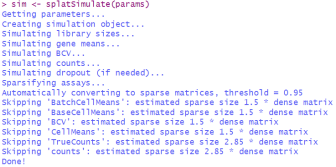
在模拟过程中,Splatter会执行一系列步骤:获取参数、创建模拟对象、模拟库大小、基因均值、生物学变异系数(BCV)、计数、如果需要的话模拟dropout(数据丢失),以及将结果稀疏化。这个过程会生成一个稀疏矩阵,这有助于我们更有效地处理和分析数据。
简单来说,我们首先从一个数据集中估计出模拟所需的参数,然后用这些参数来创建一个新的模拟数据集。接下来的部分会详细解释这些步骤,但现在,我们只需要知道这两个主要步骤就可以了。
二、Splat模拟:数据生成的奥秘
小伙伴们,在深入研究参数估算之前,先跟小师妹快速了解一下Splatter包的Splat模拟是怎么回事。Splat其实就是Splatter包里用来生成模拟数据的模型。它用伽马-泊松分布来模拟每个基因在每个细胞中的表达计数。这个过程中,每个基因的平均表达量是从伽马分布中模拟出来的,然后用生物学变异系数来调整,以符合均值-方差的关系。Splat还能模拟那些表达量异常的基因和随机的dropout现象,每个细胞的库大小也是根据对数正态分布来模拟的,这样就能更好地匹配真实数据集。
更酷的是,Splat还能模拟不同细胞类型之间的表达差异,或者细胞分化过程中表达的连续变化。这些细节我们在后面模拟计数的部分再详细说。放心,虽然听起来有点复杂,但实际操作起来就简单多了。通过模拟数据,我们可以在不接触真实样本的情况下,测试我们的分析方法,这对于研究的前期准备来说非常有帮助。
三、SplatParams对象:模拟参数的宝库
接下来,小师妹要给大家展示的是Splatter模拟中非常核心的部分——`SplatParams`对象。这个对象就像是我们的模拟参数的小宝库,里面存储了所有模拟需要的参数。让我们来创建一个,看看它长什么样吧。
params <- newSplatParams()
params

执行上面的代码后,我们会得到一个`SplatParams`类的参数对象,里面包含了基因数量、细胞数量、随机种子等全局参数,还有批次效应、平均表达水平、库大小、表达量异常基因、组别、差异表达、BCV(生物学变异系数)、dropout(数据丢失)和路径等一大堆参数。这些参数中,有些是可以估算的,有些是固定的,还有些是可以调整的。
获取和设置参数:定制你的模拟
如果你想要查看或修改某个特定参数,比如模拟的基因数量,可以用`getParam`函数来获取,用`setParam`函数来设置新值。
# 查看模拟的基因数量
getParam(params, “nGenes”)

# 设置模拟的基因数量为5000
params <- setParam(params, “nGenes”, 5000)

如果你想一次设置多个参数,可以用`getParams`或`setParams`函数。
# 一次性设置多个参数(使用列表)
params <- setParams(params, update = list(nGenes = 8000, mean.rate = 0.5))
# 作为列表提取多个参数
getParams(params, c(“nGenes”, “mean.rate”, “mean.shape”))

# 一次性设置多个参数(使用额外的参数)
params <- setParams(params, mean.shape = 0.5, de.prob = 0.2)
Params

改变了的参数会以大写字母显示,这样我们就知道它们已经被定制过了,不再是默认值。
# 直接在创建时设置参数
params <- newSplatParams(lib.loc = 12, lib.scale = 0.6)
getParams(params, c(“lib.loc”, “lib.scale”))

通过这种方式,我们可以轻松地定制我们的模拟参数,让模拟数据更贴近我们研究的需求。是不是很方便呢?
四、参数估算:让模拟更精准
你知道么,Splatter包的`splatEstimate`函数能帮我们从含有计数数据的集合中估算出许多参数。这就像是给模拟数据装上了一个精准的指南针,让它更贴近真实世界的数据。下面,就让我们一起来看看这神奇的步骤吧!
首先,我们需要获取一个模拟的计数矩阵。这里我们用`scater`包的`mockSCE`函数来生成一个模拟的单细胞数据对象`sce`,然后从中提取计数矩阵。
# 获取模拟的计数矩阵
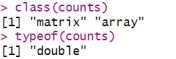
counts <- counts(sce)
# 检查counts是否为整数矩阵
class(counts)
typeof(counts)
# 查看维度,每一行代表一个基因,每一列代表一个细胞
dim(counts)
我们可以通过查看矩阵的前几行来了解数据的概貌。这个计数矩阵是一个整数矩阵,每一行代表一个基因,每一列代表一个细胞。
# 显示前几项数据
counts[1:5, 1:5]

接下来,就是重头戏——参数估算了。我们会用`splatEstimate`函数来完成这个任务。
# 参数估算
params <- splatEstimate(counts)
这个过程包括几个步骤:
1. 平均参数:通过拟合平均表达水平的伽马分布来估算。
2. 库大小参数:通过拟合库大小的对数正态分布来估算。
3. 表达量异常参数:通过确定异常值的数量,并拟合这些异常值与中位数差异的对数正态分布来估算。
4. BCV参数:使用`edgeR`包中的`estimateDisp`函数来估算。
5. Dropout参数:通过检查dropout现象是否存在,并拟合平均表达水平与零值比例之间的关系的逻辑函数来估算。
通过这些步骤,我们就能获得一组贴近真实数据的参数,用它们来模拟新的数据集,是不是很棒呢?这样一来,我们就能在不接触真实样本的情况下,测试和验证我们的分析方法了。
五、模拟计数:用`splatSimulate`生成数据
当我们对一组参数感到满意后,就可以用这些参数来生成新的数据集了。如果我们想要微调这些参数,可以直接在函数中提供它们;如果我们不提供任何参数,就会使用默认值。
sim <- splatSimulate(params, nGenes = 1000)
sim
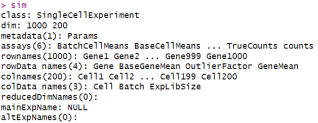
执行上面的代码后,我们会得到一个`SingleCellExperiment`对象,里面包含了1000个特征(基因)和200个样本(细胞)。这个对象的主要部分是一个特征乘以样本的矩阵,包含了模拟的计数数据,我们可以通过`counts`来访问这些数据。此外,这个对象还可以包含其他表达量度量,比如FPKM或TPM。此外,`SingleCellExperiment`对象还包含了每个细胞的表型信息(通过`colData`访问)和每个基因的特征信息(通过`rowData`访问)。Splatter利用这些插槽以及`assays`来存储模拟过程中的中间值。
# 访问计数数据
counts(sim)[1:5, 1:5]

我们可以访问关于基因的信息,比如基础基因均值、异常因子和基因均值等。
# 基因信息
head(rowData(sim))
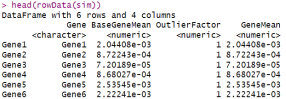
同样,我们也可以获取关于细胞的信息,比如每个细胞所属的组或路径、预期的库大小等。
# 细胞信息
head(colData(sim))
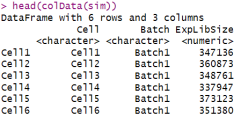
此外,我们还可以查看基因与细胞之间的矩阵信息,比如每个基因在每个细胞中的表达均值、生物学变异系数(BCV)、细胞均值、真实计数和dropout信息等。
# 基因与细胞矩阵信息
names(assays(sim))

我们还可以得到细胞均值矩阵,并查看前几行和前几列的数据:
assays(sim)$CellMeans[1:5, 1:5]
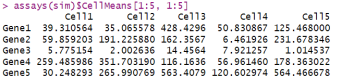
执行这个命令后,我们会得到一个矩阵,里面包含了五个基因(Gene1到Gene5)在五个细胞(Cell1到Cell5)中的表达均值。这些数值可以帮助我们理解基因在不同细胞中的表达模式。
使用`SingleCellExperiment`对象的另一个大好处是,我们可以立即使用其他分析包,比如`scater`包中的绘图函数。例如,我们可以制作PCA图来可视化数据。
# 使用scater计算logcounts并绘制PCA图
sim <- logNormCounts(sim)
sim <- runPCA(sim)
plotPCA(sim)
注意:由于模拟是随机的,每次运行的结果可能会有所不同。
通过这些步骤,我们就能生成一个包含丰富信息的模拟数据集,并且可以利用现有的分析工具进行进一步的分析。是不是感觉Splatter包超级强大呢?如果你在模拟数据的过程中有任何疑问,记得找小师妹帮忙哦!
模拟细胞群组
想象一下,我们不仅想研究单一类型的细胞,还想看看不同细胞类型混合在一起时会发生什么。Splatter包可以通过改变`method`参数来模拟这种情况。比如,我们可以设置`group.prob`参数,并把`method`参数设为”groups”来模拟两组细胞。
sim.groups <- splatSimulate(
group.prob = c(0.5, 0.5),
method = “groups”,
verbose = FALSE)
sim.groups <- logNormCounts(sim.groups)
sim.groups <- runPCA(sim.groups)
plotPCA(sim.groups, colour_by = “Group”)
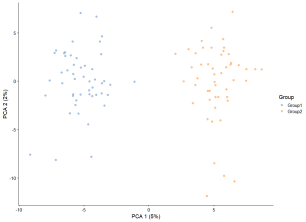
如果我们把两组细胞的概率都设为0.5,那么我们应该能得到大约相等数量的细胞(在这个例子中大约是50个)。如果我们想要不等数量的细胞群,我们可以把`group.prob`设为任何加起来等于1的概率集合。
模拟分化路径
另一个我们经常感兴趣的情况是细胞分化过程,其中一个细胞类型逐渐转变为另一种类型。Splatter通过模拟两组之间的一系列步骤,并随机将每个细胞分配给一个步骤来近似这个过程。我们可以使用”paths”方法来创建这种模拟。
sim.paths <- splatSimulate(
de.prob = 0.2,
nGenes = 1000,
method = “paths”,
verbose = FALSE)
sim.paths <- logNormCounts(sim.paths)
sim.paths <- runPCA(sim.paths)
plotPCA(sim.paths, colour_by = “Step”)
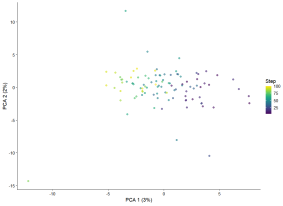
在这里,颜色代表了每个细胞在分化路径上的“步骤”。我们可以看到,颜色较深的细胞更接近原始细胞类型,而颜色较浅的细胞更接近最终分化的细胞类型。通过设置额外的参数,我们可以模拟更复杂的过程(例如,从单一祖细胞分化出多种成熟细胞类型)。
批次效应
在任何测序实验的分析中,批次效应都是一个重要因素,这是同一时间处理的一组样本中常见的技术变异。我们可以通过告诉Splatter每个批次中有多少个细胞来应用批次效应。
sim.batches <- splatSimulate(batchCells = c(50, 50), verbose = FALSE)
sim.batches <- logNormCounts(sim.batches)
sim.batches <- runPCA(sim.batches)
plotPCA(sim.batches, colour_by = “Batch”)
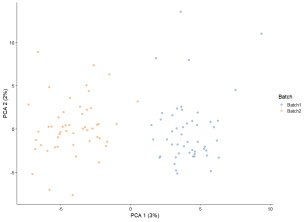
这看起来和我们模拟群组时很像,因为过程非常相似。不同的是,批次效应适用于所有基因,不仅仅是那些差异表达的基因,而且效应通常较小。通过结合群组和批次,我们可以模拟我们不感兴趣的变异(批次)和我们感兴趣的变异(群组)。
模拟不同的表达值
Splatter默认是设计来模拟计数数据的,但有些分析方法可能需要其他类型的表达值,尤其是长度标准化的值,比如TPM或FPKM。scater包有函数可以将这些值添加到SingleCellExperiment对象中,但它们需要每个基因的长度。这时,我们可以用addGeneLengths函数来模拟这些长度。
sim <- simpleSimulate(verbose = FALSE)
sim <- addGeneLengths(sim)
head(rowData(sim))

然后,我们就可以用scater来计算TPM了。
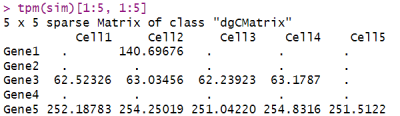
tpm(sim) <- calculateTPM(sim, rowData(sim)$Length)
tpm(sim)[1:5, 1:5]
默认情况下,addGeneLengths使用对数正态分布生成值,然后四舍五入得到整数长度。这个分布的参数是基于人类蛋白质编码基因的,但如果需要,也可以调整(比如针对其他物种)。或者,也可以从一个提供的向量中抽样长度。
缩减模拟大小
在使用Splatter包模拟数据时,我们会得到很多中间值,这些值虽然有助于评估,但也会大大增加数据的大小。如果你不需要这些中间值,我们可以用`minimiseSCE()`函数来缩减模拟数据的大小,非常实用哦!
首先,我们运行`splatSimulate()`函数来生成模拟数据:
sim <- splatSimulate()
然后,我们使用`minimiseSCE()`函数来缩减数据大小:
minimiseSCE(sim)
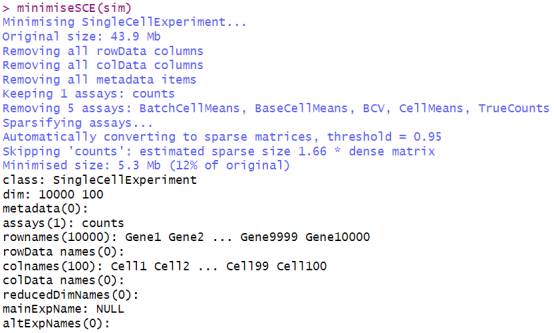
这个函数默认会移除`rowData(sce)`、`colData(sce)`和`metadata(sce)`中的所有内容,只保留`counts`。如果你有其他想要保留的内容,可以在函数中指定。例如,我们可以保留`Gene`、`Cell`和`Batch`列:
minimiseSCE(sim,
rowData.keep = “Gene”,
colData.keep = c(“Cell”, “Batch”),
metadata.keep = TRUE)
这样,我们就能得到一个更小的`SingleCellExperiment`对象,同时保留了我们关心的信息。是不是很方便呢?
六、比较模拟数据与真实数据
在模拟数据生成之后,你可能想要将其与真实数据集进行比较,或者比较不同参数或模型下的模拟结果。`splat`包提供了一个名为`compareSCEs`的函数,旨在简化这些比较工作。顾名思义,这个函数接收一组`SingleCellExperiment`对象,将数据集合并,并生成一些比较它们的图表。让我们进行两个小的模拟,看看它们如何比较。
sim1 <- splatSimulate(nGenes = 1000, batchCells = 20, verbose = FALSE)
sim2 <- simpleSimulate(nGenes = 1000, nCells = 20, verbose = FALSE)
comparison <- compareSCEs(list(Splat = sim1, Simple = sim2))
names(comparison)
names(comparison$Plots)

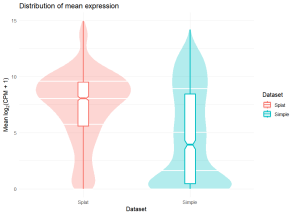
返回的列表包含三个项目。前两个是通过基因(RowData)和通过细胞(ColData)合并的数据集,第三个包含一些比较图表(使用`ggplot2`生成),例如,一个展示均值分布的图表:
comparison$Plots$Means
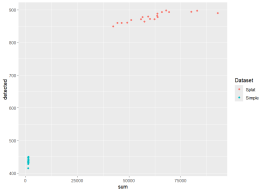
这些只是你可能想要考虑的一些图表,但应该很容易使用返回的数据制作更多图表。例如,我们可以绘制表达基因数量与库大小的关系图:
library(“ggplot2”)
ggplot(comparison$ColData, aes(x = sum, y = detected, colour = Dataset)) +
geom_point()
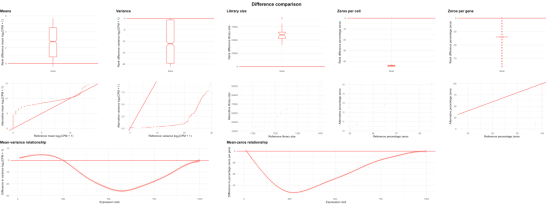
比较差异
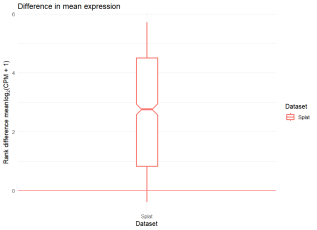
有时,与其直观地比较数据集,不如看看它们之间的差异。我们可以使用`diffSCEs`函数来实现这一点。类似于`compareSCEs`,这个函数也接收一组`SingleCellExperiment`对象,但现在我们还需要指定一个作为参考的数据集。返回的一系列图表与之前类似,但它们不是展示整体分布,而是展示与参考数据集的差异。
difference <- diffSCEs(list(Splat = sim1, Simple = sim2), ref = “Simple”)
difference$Plots$Means
我们还会得到一系列分位数-分位数图,可以用来比较分布。
difference$QQPlots$Means

制作面板
这些比较会生成多个图表,可能会让人眼花缭乱。为了简化这个过程,或者为了制作出版物中的图表,你可以使用`makeCompPanel`、`makeDiffPanel`和`makeOverallPanel`函数。
这些函数使用`cowplot`包将图表合并成一个面板。面板可能会很大,难以查看(例如,在RStudio的图表查看器中),因此最好输出面板并单独查看。幸运的是,`cowplot`提供了一个方便的函数来保存图像。以下是输出每个面板的一些建议参数:
panel <- makeCompPanel(comparison)
cowplot::save_plot(“comp_panel.png”, panel, nrow = 4, ncol = 3)
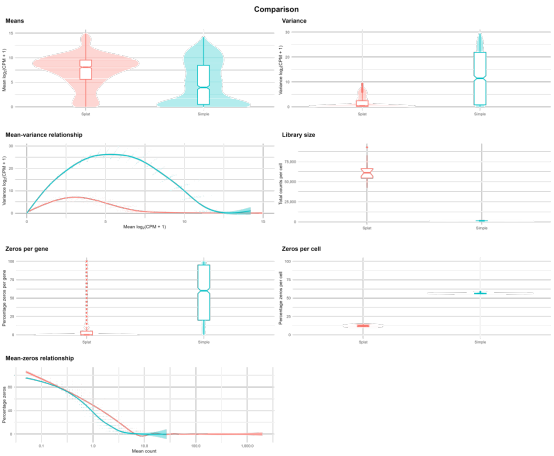
panel <- makeDiffPanel(difference)
cowplot::save_plot(“diff_panel.png”, panel, nrow = 3, ncol = 5)
panel <- makeOverallPanel(comparison, difference)
cowplot::save_plot(“overall_panel.png”, panel, ncol = 4, nrow = 7)
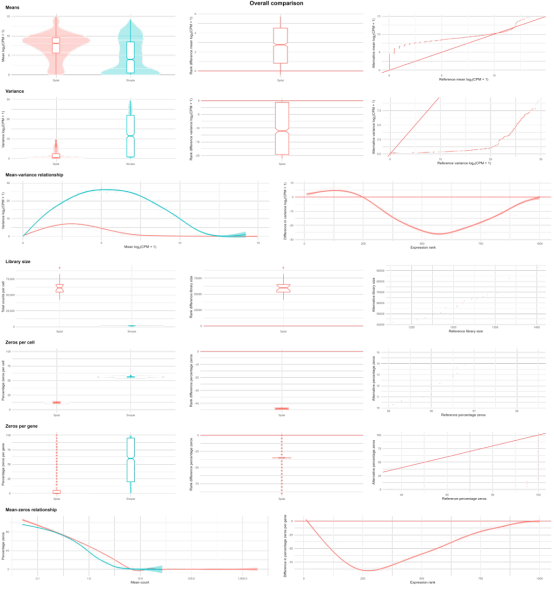
通过这些函数,我们可以更直观地比较不同模拟数据集之间的差异,并且能够将这些比较整合成一个易于查看和理解的面板。这样,无论是在研究中还是在准备发表的论文中,我们都能够更有效地展示我们的发现。
通过使用Splatter包进行模拟,我们能够高效地生成单细胞RNA测序数据,为研究提供了强有力的支持。这不仅帮助我们理解基因表达的复杂性,也为未来的生物医学研究奠定了基础。如果你对如何应用这个强大的工具有任何疑问,记得找小师妹,让我们一起探索基因组的奥秘!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~