
Hi!小师妹今天来教大家一个探索基因组信息的得力助手工具,在这个基因组学领域飞速发展的黄金时代,我们经常需要分析和比较大量的高通量测序数据,比如ChIP-seq、RNA-seq等,以揭示基因表达的奥秘和调控机制,在此之前,我们还需要将原始数据进行质控,才能顺利进行下游分析!本次介绍的软件操作占用内存比较大,建议使用服务器,欢迎联系小师妹租赁性价比居高的服务器~

很多朋友在做项目的时候,得到原始数据之后,我们可能需要先了解一下测序数据的测序深度和覆盖率等情况,才能确定数据是否需要重新测序。这个时候就需要用到这么一款神器了,其堪称“深海利器”。Deeptools2 软件在 2016 年发表在 NAR(Nucleic Acids Research)上,16 年推出的 Deeptools2 在计算和可视化方面做出了很多更新和升级,旨在更快速和便捷的分析和可视化高通量测序数据,其引用量已达3200多次,是一个靠谱、可信的质检好工具。今天小师妹给大家带来Deeptools工具的基本功能和用途,以及如何实战应用,感兴趣的话就一起看下去吧。小师妹可是厉害的生信高手,要是同学们有自己做不了的蛋白质结构分析,欢迎随时联系我!!
一、什么是Deeptools
Deeptools常用于以下任务:绘制热图、绘制平均曲线、计算基因组覆盖度和富集分析等。它还可以与常见的基因组浏览器(如IGV和UCSC Genome Browser)集成,使用户可以方便地查看和比较不同的测序数据。
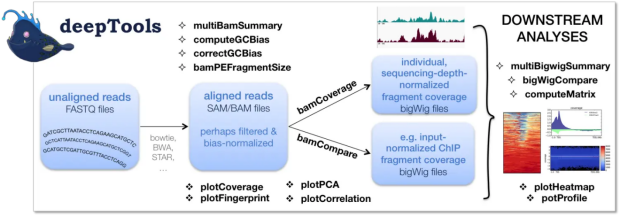
图1 工具功能示意图
二、下载和安装
作者在官网中,共推荐了4种下载方法,这里小师妹给大家带来3种最常见的下载方法:
1.Conda下载
安装DeepTool的推荐方式是通过miniconda或anaconda,前提是conda配置了国内镜像源。
$ conda install -c bioconda deeptools
2.Pip下载
Deeptools是python包,可以使用pip工具进行下载:
$ pip install deeptools
$ pip install deeptools==3.5.3 # 可以安装指定版本
3.Git下载
$ pip install deeptools==3.5.3 # 可以安装指定版本
$ git clone https://github.com/deeptools/deepTools.git
$ cd deepTools
$ pip install .
三、常见命令
1.bamCoverage
现在假设我得到了一个bam文件,应该如何生成一个可以在基因组浏览器(例如IGV)中查看的文件呢?当然,我们也可以在基因组浏览器中查看BAM文件,但直接把bam导入IGV看峰图太慢,把bam转为bigWig格式后,文件会小很多,载入也很快了。
$ bamCoverage –bam, -b <your.bam> \ # 要处理的bam文件
–outFileName, -o <output.bw> \ # 要输出的文件名
–region, -r <chr10> \ # 想要处理的区域,格式为chr:start:end
–numberOfProcessors, -p <1> \ # 线程数,默认为1
–verbose, -v \ # 显示处理过程
–binSize 10 # bin越小,分辨率越高,默认为50
然后再将bigwig文件导入IGV,记得要导入参考基因组文件,可以得到一个各样本总体表达量情况,还可以点进不同的染色体进一步查看,效果图如下。怎么样,是不是很直观呢,我们还可以调整要显示的染色体。
图2 bigwig文件导入IGV
2.plotCoverage
该工具可用于评估给定样本的测序深度。它采样100万个bp,计算重叠读取的数量,并可以报告一个直方图,告诉你有多少个碱基被覆盖了多少次。可以接受多个BAM文件,但它们都应该对应于相同的基因组组装。
$ plotCoverage -b <sample1.bam sample2.bam> \ # 空格间隔的bam文件
–plotFile, -o <coverage.png> \ # 输出文件名
–labels, -l <label1 label2> \ # 定义样本的标签,空格间隔
–plotTitle, -T <yourTitle> \ # 绘图标题
–skipZeros \ # 排除0或nan的基因组
–outRawCounts readCounts.tab\ # 将原始计数 (coverage) 保存到文件
–region, -r \ # 想要处理的区域,格式为chr:start:end
–numberOfProcessors, -p \ # 线程数,默认为1
$ head readCounts.tab
#’chr’ ‘start’ ‘end’ ‘sample1.bam’ ‘sample2.bam’ ‘sample3.bam’ ‘sample4.bam’ ‘sample5.bam’ ‘sample6.bam’
NC_072815.1 979324 979325 456.0 743.0 645.0 708.0 409.0 389.0
NC_072815.1 979336 979337 468.0 743.0 636.0 709.0 408.0 381.0
NC_072815.1 979348 979349 487.0 720.0 628.0 716.0 425.0 383.0
NC_072815.1 979360 979361 498.0 719.0 622.0 740.0 425.0 375.0
NC_072815.1 979372 979373 503.0 712.0 614.0 735.0 444.0 368.0
NC_072815.1 979384 979385 512.0 740.0 662.0 758.0 456.0 373.0
NC_072815.1 979396 979397 501.0 724.0 665.0 762.0 465.0 370.0
NC_072815.1 979408 979409 509.0 730.0 659.0 773.0 456.0 367.0
plotCoverage可以得到一个bam文件的可视化图,左图可以得到各样本平均深度,右图可以知道各深度所占比例,例如在棕色线代表的样本中,400X深度的片段占20%。
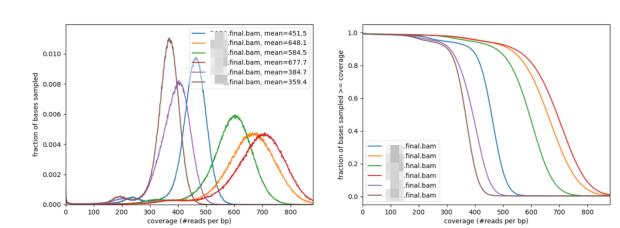
图3 plotCoverage得到平均深度和所占比例
3.computeMatrix:聚类分析
computeMatrix主要用于计算基因组区域的得分,并生成一个中间文件,该文件可以与plotHeatmap和plotProfile工具一起使用进行可视化。computeMatrix有两种主要的使用模式,原理如下图:
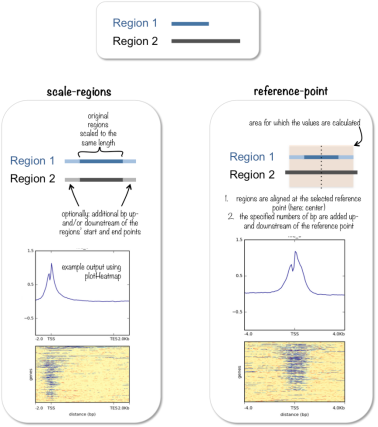
图4 computeMatrix聚类分析原理示意图
①reference-point模式:计算相对于某个点(例如基因的转录起始位点TSS、终止位点TES或区域中心)的信号分布。用户可以通过-referencePoint参数指定参考点,以及通过b和a参数定义感兴趣区域的上游和下游长度。
$ computeMatrix scale-regions -S H3K27Me3-input.bigWig \
H3K4Me1-Input.bigWig \
H3K4Me3-Input.bigWig \
-R genes19.bed genesX.bed \
–beforeRegionStartLength 3000 \
–regionBodyLength 5000 \
–afterRegionStartLength 3000
–skipZeros -o matrix_1.mat.gz
$ plotHeatmap -m matrix_2.mat.gz \
-out ExampleHeatmap2.png \
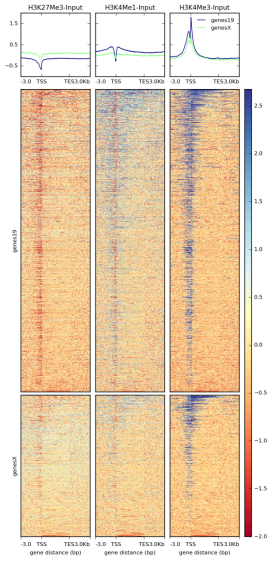
图5 reference-point用于计算相对于点(参考点)的信号分布
②scale-regions模式:计算一组区域上的信号,这些区域可以被缩放到相同的大小。用户可以通过b(beforeRegionStartLength)、a(afterRegionStartLength)和m(regionBodyLength)参数控制区域长度。
$ computeMatrix scale-regions -S H3K27Me3-input.bigWig \
H3K4Me1-Input.bigWig \
H3K4Me3-Input.bigWig \
-R genes19.bed genesX.bed \
–beforeRegionStartLength 3000 \
–regionBodyLength 5000 \
–afterRegionStartLength 3000
–skipZeros -o matrix.mat.gz
$ plotProfile -m matrix.mat.gz \
-out ExampleProfile1.png \
–numPlotsPerRow 2 \
–plotTitle “Test data profile”
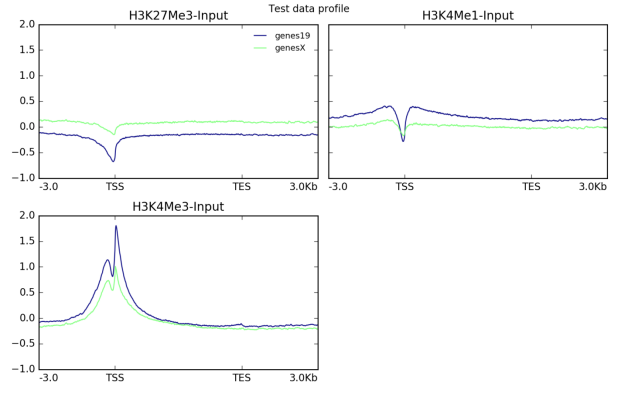
图6 scale-regions用于在一组区域(比例尺-区域)上计算信号,其中所有区域都缩放到相同的大小
4.multiBigwigSummary
multiBigwigSummary用于计算一组bigWig文件在基因组中的指定区域或整个基因组上的覆盖度平均值,这个工具有两种模式:bins和BED-file。计算2个以上的bam的基因组区域的read覆盖度。可以使用“bins”模式对整个基因组进行分析,也可以使用BED文件模式计算特定区域的reads覆盖度。
plotCorrelation根据基因组区域内的read覆盖率(或其他分数),计算两个或多个文件之间的总体相似度,必须使用multiBamSummary或multiBigwigSummary计算。Pearson是适用于符合正态分布的数据的一种适当的测量方法,而Spearman则不做出这种假设,通常不受异常值的影响,但也因此变得不太敏感。
# bins模式
$ multiBigwigSummary bins \
-b testFiles/H3K4Me1.bigWig testFiles/H3K4Me3.bigWig testFiles/H3K27Me3.bigWig testFiles/Input.bigWig \
–labels H3K4me1 H3K4me3 H3K27me3 input \
-out scores_per_bin.npz –outRawCounts scores_per_bin.tab
$ head scores_per_bin.tab
#’chr’ ‘start’ ‘end’ ‘H3K4me1’ ‘H3K4me3’ ‘H3K27me3’ ‘input’
19 0 10000 0.0 0.0 0.0 0.0
19 10000 20000 0.0 0.0 0.0 0.0
19 20000 30000 0.0 0.0 0.0 0.0
19 30000 40000 0.0 0.0 0.0 0.0
19 40000 50000 0.0 0.0 0.0 0.0
19 50000 60000 0.0221538461538 0.0 0.00482142857143 0.0522717391304
19 60000 70000 4.27391282051 1.625 0.634116071429 1.29124347826
19 70000 80000 13.0891675214 24.65 1.8180625 2.80073695652
19 80000 90000 1.74591965812 0.29 4.35576785714 0.92987826087
$ plotCorrelation -in matrix.gz -c spearman -p heatmap -o plot.png
# BED-file模式
$ multiBigwigSummary BED-file \
–bwfiles testFiles/*bigWig \
–BED testFiles/genes.bed \
–labels H3K27me3 H3K4me1 H3K4me3 HeK9me3 input \
-out scores_per_transcript.npz –outRawCounts scores_per_transcript.tab
$ head scores_per_transcript.tab
#’chr’ ‘start’ ‘end’ ‘H3K27me3’ ‘H3K4me1’ ‘H3K4me3’ ‘HeK9me3’ ‘input’
19 60104 70951 0.663422099099 4.37103606574 14.9609108509 0.596631607217 1.34274297191
19 60950 70966 0.714223982699 4.54650763906 16.2336261981 0.62173674295 1.41719308888
19 62114 70944 0.747578769617 4.84009060023 18.2951302378 0.648723472352 1.51324474371
19 63820 70951 0.781816722009 5.30500631048 22.5579862572 0.682862029229 1.55490104062
19 65057 66382 0.528301886792 5.45886792453 0.523018867925 0.555471698113 1.97056603774
19 65821 66416 0.411764705882 3.0 0.636974789916 0.168067226891 1.67226890756
19 65821 70945 0.844600775761 4.79176424668 31.1346604215 0.693073728066 1.47911787666
19 66319 66492 0.774566473988 1.59537572254 0.0 0.0 0.578034682081
19 66345 71535 0.877430197151 5.49036608863 43.978805395 0.746026011561 1.43545279383
multiBigwigSummary选项可以对多个bw文件进行整合,然后结合plotCorrelation和plotPCA可以实现样本相关性分析。此外,multiBamSummary还可以生成bedGraph文件,以便进行进一步的分析。
$ deepTools2.0/bin/plotCorrelation \
-in scores_per_transcript.npz \
–corMethod pearson –skipZeros \
–plotTitle “Pearson Correlation of Average Scores Per Transcript” \
–whatToPlot scatterplot \
-o scatterplot_PearsonCorr_bigwigScores.png \
–outFileCorMatrix PearsonCorr_bigwigScores.tab
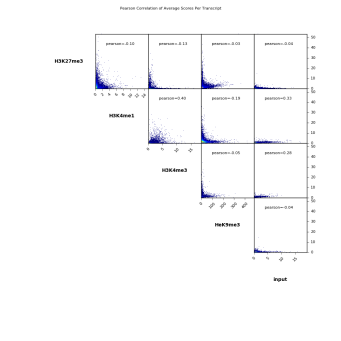
图7 使用 multiBigwigSummary 计算每个转录本的平均得分,并包括每个比较的 Pearson 相关系数,然后对其进行成对散点图
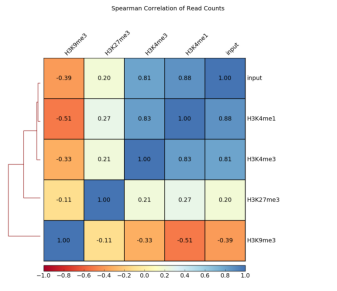
图8 生成热图,其中成对相关系数用不同的颜色强度描绘,并使用分层聚类进行聚类。
以上代码就是对Deeptools的介绍与基本使用方法,学会这些方法后,你可以学习如何使用Deeptools进行bam文件分析,包括转换为bigwig文件、获取测序覆盖率,以及对基因区域进行统计和作图等。当然,大家也可以根据自己的需求和研究目标修改和扩展这些代码哦!!怎么样,你学会了嘛!?
学完了操作流程是不是想马上操作一番,小师妹也在资料中存放了测试数据。加师妹微信获取资料哦~大家可以学习并测试一下,让知识更加牢固。
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!