
Hi!这里是小师妹。还记得本科上课时老师拿着系统进化分析忽悠你,会用MEGA吗?又或是在Windows上一通操作Mega软件,功能虽多但繁杂,而且运行速度还很受限于本地电脑的配置,今天小师妹就给大家带来删繁就简版的系统发育分析,最大程度减轻小白的烦恼,赶紧拿起小本本记好!
这个软件支持多种比对算法,包括FFT+NS+2、FFT+NS+i、FFT+NS+1、L+INS+i、G+INS+i和E+INS+i等等。重要的是,我们可以根据自己的需求选择不同的算法进行比对,而且就算你不会,模型还会帮你选择最佳模型哦~本次介绍的软件操作占用内存比较大,建议使用服务器,欢迎联系小师妹租赁性价比居高的服务器~
当然我们不能只限于对软件的使用,还要了解软件原理才能游刃有余,不被器具所支配。今天小师妹则给大家带来多序列比对神器mafft工具的分析。它不仅快速、高效、准确,支持多种操作系统,包括Windows、Linux和Mac OS等,而且可以同时处理大量的序列数据,并生成高质量的比对结果。接下来,同学们跟紧小师妹的步伐,让我们正式开启对Mafft的学习之旅吧! 如果你在操作过程中遇到任何问题,别犹豫,记得联系小师妹哦~

一、什么是Mafft?
一提到多重序列比对,很多人禁不住就想到ClustalW,而对于几千条序列的多序列比对,无论是从准确度还是运行速度上考虑,muscle通常都是最佳选择。但是muscle的内存优化做的并不好,如果所需内存超出了机器内存,此时可以考虑mafft这个工具。
MAFFT全称是Multiple Alignment using Fast Fourier Transform,起初是为了执行大规模序列比对而发展起来的,它是一种基于快速傅里叶变换的组对组的比对算法,并且使用了一种近似距离计算方法便于进行快速计算。
背景知识:傅里叶变换原理即任何一个复杂的波都可以拆解成许多个sine(wave),把一个复杂的波转化成为多个标准正弦波叠加的过程就是傅里叶变换。

图1 工具引用情况
二、下载和安装
mafft 是一个跨平台的工具,MAFFT官方网站:http://mafft.cbrc.jp/alignment/software/,支持平台:Mac OS X 、Linux、Windows。下面给大家带来linux系统的安装,需要根据自己操作系统选择相应版本下载:
图2 Mafft工具官网
第一步:下载安装包
$ wget -c https://mafft.cbrc.jp/alignment/software/mafft-7.520-linux.tgz
第二步:解压
$ tar -zxvf mafft-7.520-linux.tgz
第三步:改名
$ mv mafft-linux64 mafft
$ cd mafft
$ chmod a+x ./mafft.bat
$ mv mafft.bat mafft
三、常见命令
1.基本比对命令
输入序列的类型(氨基酸或核苷酸)是自动识别的
$ mafft sample1.fasta > sample1.output.fasta
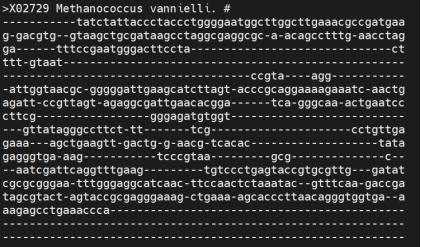
图3 比对结果文件
按照最长序列,把相应位置的aa标出来,有gap的地方,则用—补齐。
2.迭代比对
最大迭代次数–maxiterate,这里使用了–maxiterate设置最大迭代次数和–localpair比对策略进行局部比对。
$ mafft –maxiterate 1000 –localpair input.fasta > output.fasta
3.比对策略
mafft 支持核酸和蛋白序列的多序列比对,并内置了多种序列比对,这里主要介绍High accuracy中的三种比对方法:
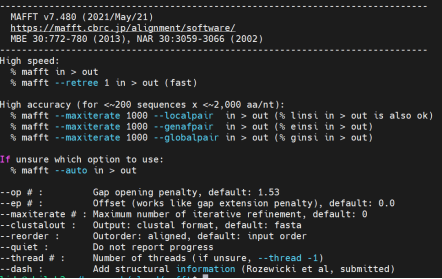
图4 Mafft帮助文档
1)L-INS-I:最准确的方法,适合小于200条,且长度小于2000aa/nt的序列。
$ mafft –maxiterate 1000 –localpair input > output
2)G-INS-I:适合序列长度相似的比对,小于200条,且长度小于2000aa/nt的序列。
$ mafft –maxiterate 1000 –globalpair input > output
3)E-INS-I:适合序列中包含较大的非匹配区域,小于200条,且长度小于2000aa/nt的序列
$ mafft –maxiterate 1000 –genafpair input > output
4.自动选择比对策略
前面讲了这么多的策略,是不是觉得有点懵了?没关系,这里还可以选择自动比对策略,使用–auto选项,MAFFT会根据输入序列的大小和数量自动选择最合适的比对策略。
$ mafft –auto input.fasta > output.fasta
5.调整线程数以加快比对速度
我们还可以使用–thread选项可以指定线程数,以并行处理来加快比对速度,但是线程数和比对速度并不是线性递增关系,存在一个最佳性能线程数,如果有批量跑多序列比对需求的伙伴,可以去了解一下。
$ mafft –thread 24 input.fasta > output.fasta
四、小结
最终输出的文件格式还是fasta文件格式,至此,就完成了mafft多序列比对。后续我们可以将结果文件导入到mega进行可视化和修剪,导出成phy格式,即可进行系统发育树的构建啦。
图5 对序列比对文件导入Mega软件进行可视化
如果小伙伴用的是自己的台式电脑,不能进行大任务量的多序列比对,EBI也提供了mafft的在线服务,网址如下:
图6 EBI官网Mafft在线工具
当然,市面上还有很多其他的多序列比对软件,有文献对多种多序列比对工具进行了比较:比对速度Muscle>MAFFT>ClustalW>T-Coffee,比对准确性MAFFT>Muscle>T-Coffee>ClustalW。因此,推荐使用 MAFFT 软件进行多序列比对。
总的来说,MAFFT 适合大规模基因组项目、多样化的数据集比对,以及需要灵活算法选择的研究,由于参数众多,新手可能需要时间学习如何有效使用,不过我们也可以“傻瓜式”选择auto参数,一键解决所有烦恼~
既然说到建树基于多序列比对,就不得不介绍一下整体流程:
①挑选合适的基因生成fasta文件(DNA/AA)
②多序列比对 (Mafft/ ClustalW)
③去掉比对中的gap (TrimAl)
④转换格式,生成的FASTA, NEXUS, CLUSTALW格式的文件均
⑤建树 (IQ-TREE)
⑥进化树美化(figtree、itol、mega)
今天小师妹给大家带来的是第二步的软件mafft分享,关注小师妹,后续还会继续带来系统发育分析的分享哦~
同学们如果觉得自己写代码麻烦,可以体验一下我们的云生信小工具,只需输入数据,即可轻松生成所需图表。立即访问云生信(http://www.biocloudservice.com/home.html),开启便捷的生信之旅!