面对复杂的RNA-Seq数据,你是不是也曾感到无从下手呢?别担心,今天,小师妹要给你们展示一个超给力的R包——EDASeq。有了它,这一切都将变得简单起来。接下来,我们会一起学习如何对RNA测序实验数据进行探索性数据分析(EDA)和标准化处理。
你知道吗?在RNA测序的世界里,我们通常把EDA看作是两步走的战略:先是读段级别的EDA,它能帮助我们发现测序深度低、质量有问题的通道,以及那些不寻常的核苷酸频率。然后是基因级别的EDA,它能让我们捕捉到错误标记的通道、分布假设问题(比如过度离散)和GC含量偏差。
EDASeq包不仅提供了“单通道”和“多通道”的标准化程序,来分别处理通道内基因特异性(可能还有通道特异性)的读数影响,比如与基因长度或GC含量有关的因素,还有处理通道间读数分布差异的影响,比如测序深度。
要注意的是,EDASeq在处理大量数据时可能会需要比较大的内存,所以小师妹建议你们用服务器来跑这些分析。如果你们需要租用服务器,记得找小师妹,给你们安排得明明白白。
好了,现在你们准备好了吗?接下来,就让我们从安装包开始,一步步走进EDASeq的世界。遇到任何问题,小师妹都会在这里,手把手教你们怎么用。一起加油,揭开RNA-Seq数据的所有秘密吧!
首先,让我们开始安装EDASeq包和另外两个需要用到的包:
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“EDASeq”)
BiocManager::install(“yeastRNASeq”)
BiocManager::install(“leeBamViews”)
安装好了之后,记得加载这些必要的库:
library(EDASeq)
library(yeastRNASeq)
library(leeBamViews)
现在,我们已经全副武装,准备开始这场激动人心的数据分析之旅了!
一、 读取未比对和已比对的测序数据
- 未比对的测序数据
存储在FASTQ格式中的未比对(未映射)测序数据可以通过从ShortRead包中导入的FastqFileList类来管理。可以在FastqFileList对象的elementMetadata槽中存储与每个通道中序列化的库有关的信息。
files <- list.files(file.path(system.file(package = “yeastRNASeq”), “reads”), pattern = “fastq”, full.names = TRUE)
names(files) <- gsub(“ll.fastq.*”, “”, basename(files))
met <- DataFrame(conditions = c(rep(“mut”, 2), rep(“wt”, 2)), row.names = names(files))
fastq <- FastqFileList(files)
elementMetadata(fastq) <- met
除了这种方法,你也可以直接加载小师妹准备好的数据文件哦~
fastq <- readRDS(file = “fastq_data.rds”)

- 已比对的测序数据
该包可以处理BAM格式的已比对(已映射)测序数据,使用的是Rsamtools包中的BamFileList类。同样,elementMetadata槽可以用来存储通道级别的样本信息。
files <- list.files(file.path(system.file(package = “leeBamViews”), pattern = “bams”, full.names = TRUE))
names(files) <- gsub(“.bam”, “”, basename(files))
gt <- gsub(“*/”, “-“, files)
gt <- gsub(“_.*”, “”, gt)
lane <- gsub(“.*(.)s”, “ll”, gt)
geno <- gsub(“.$”, “”, gt)
pd <- DataFrame(geno = geno, lane = lane, row.names = paste(geno, lane, sep = “.”))
bfs <- BamFileList(files)
elementMetadata(bfs) <- pd
除了这种方法,你也可以直接加载小师妹准备好的数据文件哦~
bfs <- readRDS(file = “bfs_data.rds”)

二、读段级别的EDA
- 未比对和已比对读段的数量
进行质量控制的一个重要检查是查看每个通道产生的总读段数,以及已映射到参考基因组的读段数和百分比。总读段数低可能表示输入RNA的质量差,而映射百分比低可能表明读段质量差(复杂性低)、参考基因组的问题或通道被错误标记。
colors <- c(rep(rgb(1, 0, 0, alpha = 0.7), 2), rep(rgb(0, 0, 1, alpha = 0.7), 2), rep(rgb(0, 1, 0, alpha = 0.7), 2), rep(rgb(1, 1, 0, alpha = 0.7), 2))
barplot(bfs, las = 2, col = colors)
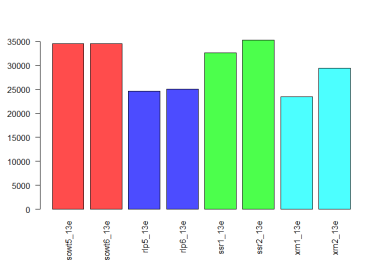
使用BamFileList类的barplot方法生成的图显示了包含在包`leeBamViews`中的酵母数据集的子集的已映射读段数。遗憾的是,`leeBamViews`没有提供未比对的读段,但是可以使用FastqFileList类的barplot方法获得总读段数的条形图。类似地,可以使用带有签名`c(x = “BamFileList”, y = “FastqFileList”)`的plot方法绘制已映射读段的百分比。
- 读段质量得分
作为额外的质量检查,可以绘制每个通道中未映射或已映射读段的平均每个碱基(即每个周期)的质量得分。
plotQuality(bfs, col = colors, lty = 1)
legend(“topright”,unique(elementMetadata(bfs)[,1]), fill=unique(colors))
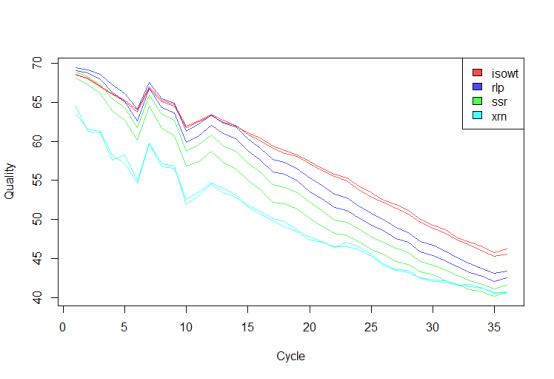
- 单独通道的总结
如果对某个通道更感兴趣,可以更详细地查看每个通道的每个碱基的质量得分分布,以及按染色体或链分隔的已映射读段数。正如预期的那样,所有读段都映射到第XII染色体。
plotQuality(bfs[[1]], cex.axis = 0.8)
barplot(bfs[[1]], las = 2)


- 读段核苷酸分布
与序列组成相关的潜在偏差源。`plotNtFrequency`函数绘制给定通道中所有读段的每个碱基的核苷酸频率。
plotNtFrequency(bfs[[1]])

上述内容包括了如何进行读段级别的探索性数据分析(EDA),以及如何使用R代码来检查读段的质量。
三、 基因级别的EDA
- 基因级别计数的类别和方法
基因级别的EDA同样重要,因为有时一些偏差在较小的尺度上难以检测。以下是如何使用`EDASeq`包来处理基因级别的计数数据。
首先,我们加载基因级别的计数数据:
data(geneLevelData)
head(geneLevelData)

然后,我们加载基因长度和GC含量信息:
data(yeastGC)
head(yeastGC)

data(yeastLength)
head(yeastLength)

接下来,我们过滤掉非表达的基因,即只考虑那些在四个通道中平均读数计数超过10的基因,并且我们有它们的长度和GC含量信息。这给我们留下了4994个基因。
filter <- apply(geneLevelData,1,function(x) mean(x)>10)
table(filter)

common <- intersect(names(yeastGC),rownames(geneLevelData[filter,]))
length(common)

EDASeq包提供了SeqExpressionSet类来存储基因计数、通道级别的信息以及基因级别的特征信息。我们使用之前创建的数据框met作为通道级别的数据。至于特征数据,我们使用基因长度和GC含量。
feature <- data.frame(gc=yeastGC,length=yeastLength)
data <- newSeqExpressionSet(counts=as.matrix(geneLevelData[common,]),
featureData=feature[common,],
phenoData=data.frame(
conditions=factor(c(rep(“mut”,2),rep(“wt”,2))),
row.names=colnames(geneLevelData)))
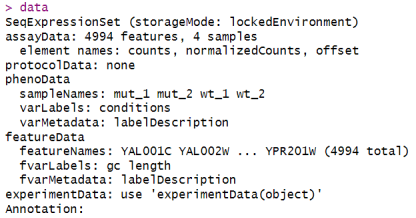
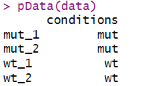
请注意,计数和特征数据的行名必须相同;同样,phenoData的行名和计数的列名也必须相同。表达值可以通过counts访问,通道信息可以通过pData访问,特征信息可以通过fData访问。
head(counts(data))

pData(data)
head(fData(data))
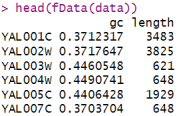
SeqExpressionSet类有两个额外的槽:normalizedCounts和offset(与计数矩阵相同维度),这些可以用来存储后续分析中使用的标准化计数矩阵和标准化偏移量。如果没有指定,偏移量将初始化为零矩阵。
head(offset(data))
- 基因级别计数的通道间分布
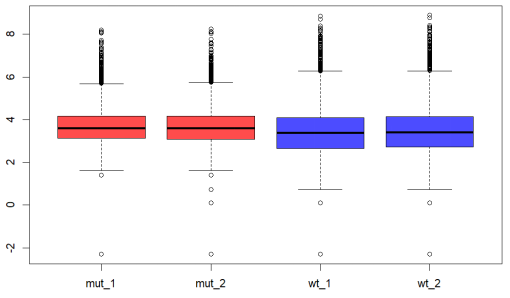
处理基因级别计数数据时,一个主要考虑因素是通道间计数分布的差异。`boxplot`方法提供了一种简单的方式,可以生成每个通道中基因计数对数的箱线图。
boxplot(data, col = colors[1:4])
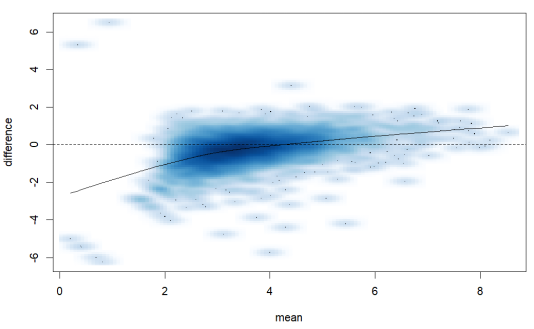
MDPlot方法可以生成两个通道的计数平均差异图(MD-plot)。中心线周围的点表示基因表达相似。远离中心线的点可能表示重要的差异表达基因。
MDPlot(data,c(1,3))
- 过度离散
尽管泊松分布是模拟计数数据的一种自然且简单的方式,但它有一个局限性,即假设均值和方差相等。因此,当数据显示过度离散时,负二项分布被提出作为替代方案。函数meanVarPlot可以用来检查计数数据是否过度离散(对于泊松分布,我们期望图中的点均匀地围绕黑色线散布)。
meanVarPlot(data[,1:2], log=TRUE, ylim=c(0,16))
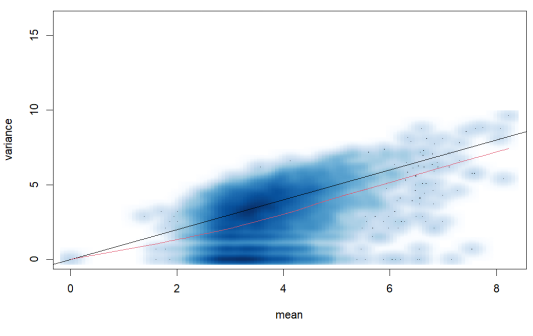
meanVarPlot(data, log=TRUE, ylim=c(0,16))
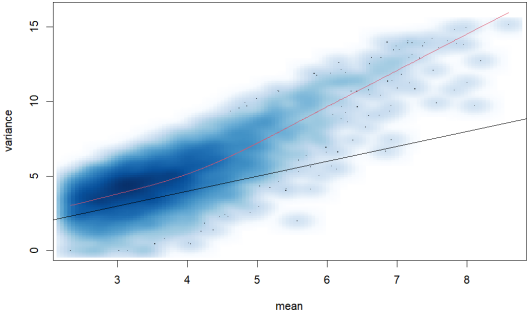
需要注意的是,均值-方差关系应该仅在重复通道内进行检查(即,在控制了预期会导致差异表达的变量的条件下)。对于酵母数据集,看到两个突变型技术重复通道没有过度离散的证据并不奇怪;同样地,两个野生型通道也是如此。然而,当同时考虑所有四个突变型和野生型通道时,考虑到生物学变异的存在,预计会看到过度离散现象。
- 基因特异性对读数计数的影响
多位作者已经报告了与基因长度、GC含量和可映射性等序列特征相关的选择偏差(Bullard et al. 2010, @hansen2011removing, @oshlack2009transcript, @risso2011gc)。
在下面的图中,使用biasPlot获得的图形展示了基因水平计数对GC含量的依赖性。相同的图也可以为基因长度或可映射性而不是GC含量创建。
biasPlot(data, “gc”, log=TRUE, ylim=c(1,5))

为了显示GC含量依赖性可能如何影响差异表达分析,可以使用biasBoxplot方法从两个通道生成读数计数的对数倍数变化的分层箱线图。同样,可以为基因长度或可映射性创建相同类型的图。
lfc <- log(counts(data)[,3]+0.1) – log(counts(data)[,1]+0.1)
biasBoxplot(lfc, fData(data)$gc)
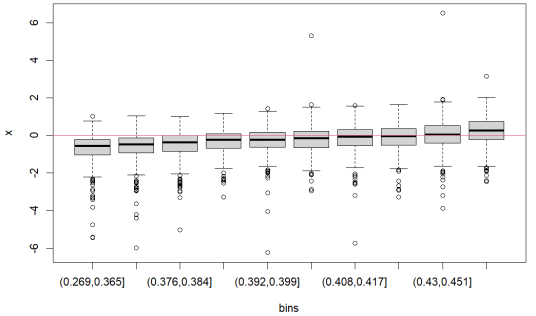
- 标准化
接下来,我们将一起看看如何使用EDASeq包进行数据标准化。标准化是RNA-Seq数据分析中一个非常关键的步骤,它可以帮助我们减少技术偏差,使得数据更适用于比较和分析。
EDASeq包实现了两种主要类型的标准化方法:within-lane(通道内)和between-lane(通道间)。Within-lane标准化考虑的是通道内基因特异性的影响,例如基因长度或GC含量;而between-lane标准化则考虑的是通道间读数分布的差异,例如测序深度。
小师妹建议在进行通道间标准化之前先对通道内效应进行标准化哦~
# 实施within-lane标准化
dataWithin <- withinLaneNormalization(data,”gc”, which=”full”)
# 实施between-lane标准化
dataNorm <- betweenLaneNormalization(dataWithin, which=”full”)
在标准化过程中,我们还可以计算偏移量(offset),它在后续的统计模型中用于校正。
通道间标准化可以用三种方法:使用中位数进行全局缩放(median)、使用上四分位数进行全局缩放(upper),以及全分位数标准化(full)。
下面的图展示了在全分位数通道内外标准化之后,GC含量偏差被减少,每个通道中的计数分布变得相同。
biasPlot(dataNorm, “gc”, log=TRUE, ylim=c(1,5))

boxplot(dataNorm, col=colors)

标准化是确保RNA-Seq数据质量的重要步骤,通过选择合适的方法和参数,我们可以有效地减少技术偏差,提高分析结果的可靠性。
伙伴们,随着小师妹的引导,我们已经完成了EDASeq的初步探索之旅。我们从读段级别的探索性数据分析入手,逐步深入到基因级别,探究了基因计数的通道间分布、过度离散现象以及基因特异性偏差。最终,我们还掌握了关键的数据标准化技术,包括通道内和通道间的标准化方法。准备好了吗?现在,你可以开始自己动手,利用EDASeq包来分析RNA-Seq数据啦!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~