嘿,科研小伙伴们!小师妹又来给大家带来一个超棒的生物信息学工具——multistateQTL包。这个工具可以让你深入探索基因如何在不同组织或环境条件下影响复杂的数量性状,从而为精准医学和个性化治疗提供科学依据。
QTL(数量性状位点)是基因组中影响数量性状的区域,而数量性状是指那些在一个群体中以连续或量化的方式表现的性状(身高、体重或者疾病易感性等)。通过研究QTL,我们可以更好地理解哪些基因变异影响这些性状,并探索它们如何在不同的生物学背景下发挥作用。
multistateQTL包就是为此而生的,它专门用来分析不同状态下的QTL汇总统计数据,比如不同组织、细胞类型或环境条件下的基因表达。想象一下,你可以轻松地对成千上万的基因、转录本或基因组区域在不同状态下的QTL数据进行统计测试、总结和可视化,这是不是听起来就很酷?
别急,小师妹现在就带你一起探索multistateQTL的神奇功能,让你的研究工作更加得心应手。注意哦,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小师妹租赁性价比高的服务器~
准备好了吗?让我们一起开启这场跨状态QTL分析的探索之旅吧!如果你在操作过程中遇到任何问题,别犹豫,记得联系小师妹哦~
首先,让我们开始安装multistateQTL包和另外需要用到的包:
if (!require(“BiocManager”, quietly=TRUE))
install.packages(“BiocManager”)
BiocManager::install(c(“QTLExperiment”, “multistateQTL”), version=”devel”)
安装好了之后,记得加载这些必要的库:
library(QTLExperiment)
library(multistateQTL)
好啦,大家准备好了吗?让我们现在就启程,一起踏上这场激动人心的数据分析之旅吧!
一、 模拟多状态QTL数据
- 从GTEx估计参数
multistateQTL 包提供了一个强大的工具,用于模拟基于真实QTL汇总统计数据的多状态QTL数据。这一过程首先需要从GTEx (v8) 公开的汇总统计数据中估计关键参数,这些参数将用于后续的数据模拟。
为了模拟具有多个生物学状态(例如不同组织)的QTL效应,我们首先使用 sumstats2qtle 函数将GTEx的汇总统计数据转换为 QTLExperiment 对象。这些数据包括了分子特征ID、变异ID、效应大小(betas)和标准误差(errors),可选地还包括p值或似然比(lfsr)。
input_path <- system.file(“extdata”, package=”multistateQTL”)
state <- c(“lung”, “thyroid”, “spleen”, “blood”)
input <- data.frame(
state=state,
path=paste0(input_path, “/GTEx_tx_”, state, “.tsv”))
gtex <- sumstats2qtle(
input,
feature_id=”molecular_trait_id”,
variant_id=”rsid”,
betas=”beta”,
errors=”se”,
pvalues=”pvalue”,
verbose=TRUE)
gtex
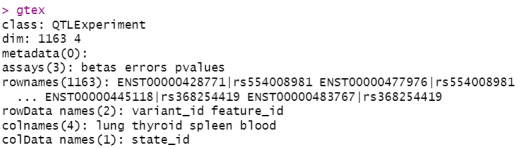
head(betas(gtex))

接下来,我们使用 qtleEstimate 函数来估计显著和非显著QTL的参数,包括形状和速率参数,这些参数定义了用于模拟效应大小和变异系数(cv)的伽马分布。
params <- qtleEstimate(gtex, threshSig=0.05, threshNull=0.5)
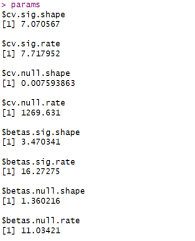
这些参数允许我们模拟出在至少一个状态下显著的QTL的平均效应大小,以及在显著和非显著状态下的QTL的cv。通过这些模拟,我们可以更深入地理解QTL在不同生物学状态下的作用模式。
为了更好地理解这些参数如何影响模拟数据,我们可以使用 plotSimulationParams 函数来可视化由这些参数定义的伽马分布。
plotSimulationParams(params=params)
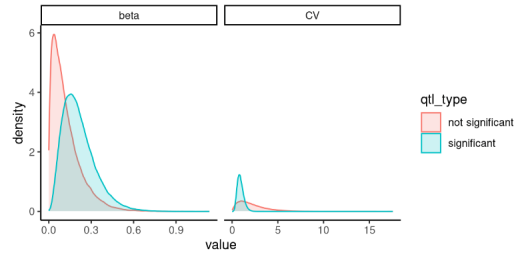
这有助于我们了解显著QTL的模拟效应大小倾向于更大,而非显著QTL的模拟cv值则相对较小。这种可视化方法为我们提供了一个直观的方式来评估模拟数据的质量和特性。
- 模拟不同类型的关联
想象一下,我们可以在电脑上模拟出基因在不同身体部位或不同环境下的作用变化,这听起来是不是很酷?multistateQTL这个工具就能帮我们做到这一点。
这个工具可以模拟出四种情况:
- 全局效应:就是基因在所有情况下都有差不多的影响。
- 独特效应:某个基因只在特定情况下起作用。
- 多状态效应:基因在几个特定情况下有影响。
- 无效应:基因在任何情况下都不发挥作用。
我们要做的就是设定一些参数,告诉电脑我们想要模拟多少基因变化,以及它们属于上述哪一种情况。然后,电脑就会随机生成一大堆数据,模拟出基因在不同情况下的作用。
# 这是一个简单的模拟示例,一半的基因变化在所有情况下都有影响
sim <- qtleSimulate(nTests=1000, nStates=6, global=0.5)
sim
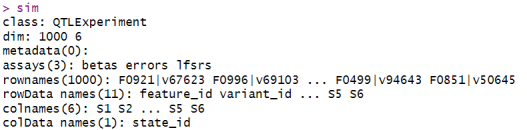
head(rowData(sim))
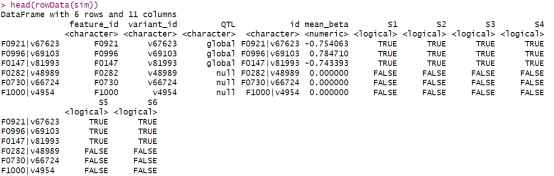
如果你想模拟更复杂的情况,比如基因变化在不同情况下的比例不同,也可以轻松做到:
# 这个模拟中,基因变化有20%是全局的,40%是多状态的,20%是独特的,20%是无影响的
sim <- qtleSimulate(
nStates=10, nFeatures=100, nTests=1000,
global=0.2, multi=0.4, unique=0.2, k=2)
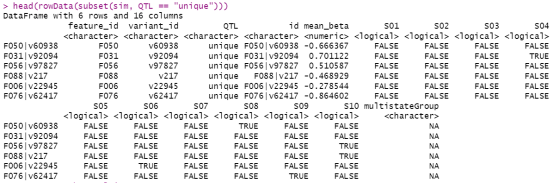
# 唯一型QTL的模拟关键指标快照
head(rowData(subset(sim, QTL == “unique”)))
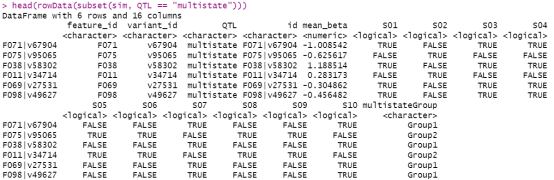
# 多状态型QTL的模拟关键指标快照
head(rowData(subset(sim, QTL == “multistate”)))
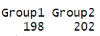
# 每个状态组特定的QTL数量
table(rowData(subset(sim, QTL == “multistate”))$multistateGroup)
通过这种方式,我们可以模拟出基因在不同环境下的作用,这对于研究基因如何影响疾病和身体功能非常有帮助。这种模拟方法为研究提供了一个灵活的工具,可以根据研究的具体需求调整模拟参数。
二、处理缺失数据
在分析过程中,缺失数据是一个常见的问题。multistateQTL工具包提供了两个实用的函数来帮助我们处理这个问题:getComplete和replaceNAs。
下面是如何在模拟数据中应用这些函数的一个示例。首先,我们人为地在模拟数据中添加了一些缺失值(NA):
na_pattern <- sample(seq(1, ncol(sim)*nrow(sim)), 1000)
sim_na <- sim
assay(sim_na, “betas”)[na_pattern] <- NA
assay(sim_na, “errors”)[na_pattern] <- NA
assay(sim_na, “lfsrs”)[na_pattern] <- NA
message(“Number of simulated tests: “, nrow(sim_na))
head(betas(sim_na))
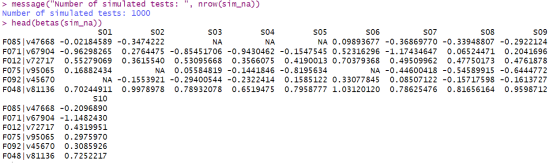
然后,我们使用getComplete函数来移除那些缺失数据过多的行,接着使用replaceNAs函数来填补剩余的缺失值。
sim_na <- getComplete(sim_na, n=0.5, verbose=TRUE)
sim_na <- replaceNAs(sim_na, verbose=TRUE)
head(betas(sim_na))

通过这些步骤,我们可以确保分析中的数据尽可能完整,从而提高分析结果的可靠性。处理缺失数据是数据分析中的一个重要环节,multistateQTL包提供的这些工具使得这个过程变得更加简单高效。
三、确定显著性
multistateQTL工具包还提供了一个callSignificance函数,它使用单一或两步阈值方法在每个状态下确定QTL测试的显著性。例如,我们可以设置一个单一的似然比(lfsr)阈值为0.1来确定模拟QTL的显著性。
sim <- callSignificance(sim, assay=”lfsrs”, thresh=0.05)
然后,我们可以计算每个状态下显著测试的中位数数量:
message(“Median number of significant tests per state: “,
median(colData(sim)$nSignificant))

因为我们有模拟的基准真值,所以我们可以使用simPerformance函数将这些显著性调用与模拟结果进行比较,该函数提供了以下全局(即跨所有状态)的性能指标:
sim <- callSignificance(sim, assay=”lfsrs”, thresh=0.001)
perf_metrics <- simPerformance(sim)
lapply(perf_metrics, FUN=function(x) {round(x, 2)})
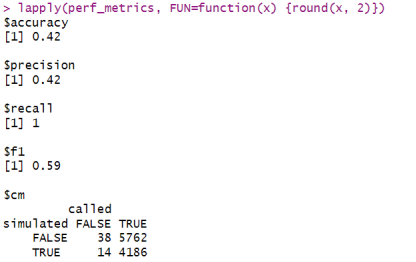
这些指标包括准确性、精确度、召回率和F1分数,以及一个混淆矩阵。从结果中我们可以看到,真正显著QTL的召回率相当低。然而,如果我们改变显著性调用方法,使其更加灵活:
sim <- callSignificance(
sim, mode=”simple”, assay=”lfsrs”,
thresh=0.0001, secondThresh=0.0002)
simPerformance(sim)$recall
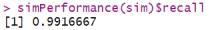
通过这种方式,我们可以显著提高真正显著QTL的召回率,从而更准确地识别出在不同状态下起作用的QTL。这对于理解基因如何在不同生物学背景下发挥作用至关重要。
四、可视化多状态eQTL数据
eQTL(Expression Quantitative Trait Loci)是指表达量数量性状位点,这是一种基因组区域,它们影响基因表达的水平。简而言之,eQTL关联研究就是为了找出哪些基因组中的位点(通常是单核苷酸多态性,即SNPs)会影响基因的表达量。这种研究有助于我们理解基因表达的遗传基础,也就是为什么在不同的个体或组织中,相同基因的表达水平会有所不同。
通过研究eQTL,科学家们可以揭示基因表达的遗传调控机制,这对于理解复杂疾病的遗传学、个体对药物反应的差异,以及生物体对环境变化的适应性都有重要意义。例如,某些eQTL可能与特定疾病的易感性或治疗反应就可能有直接的联系。因此,eQTL研究是基因组学和遗传学研究中的一个重要领域。
multistateQTL 包提供了五个函数来可视化多状态eQTL数据。这些函数基于 ggplot2 和 ComplexHeatmap R包构建。这五个函数分别是plotPairwiseSharing、plotQTLClusters、plotUpSet、plotCompareStates和plotSimulationParams。
- 成对共享
plotPairwiseSharing 函数展示了每对状态之间显著命中的成对共享程度。可以通过指定对象的 colData 中的有效列名来添加列注释。
在下面的图中,列根据状态的广泛细胞类型(multistateGroup)排序。我们期望属于同一多状态组的状态更加相关,并且在显著 eQTLs 上有更高的共享程度。列注释用于显示每个状态的显著 eQTL 数量。
sim_sig <- getSignificant(sim)
sim_top <- getTopHits(sim_sig, assay=”lfsrs”, mode=”state”)
sim_top <- runPairwiseSharing(sim_top)
p1 <- plotPairwiseSharing(sim_top, annotate_cols=c(“nSignificant”, “multistateGroup”))
- UpSet 图
下图展示了由不同状态组显著但不必然共享的测试集。
plotUpSet(sim_top, annotateColsBy=c(“nSignificant”, “multistateGroup”))
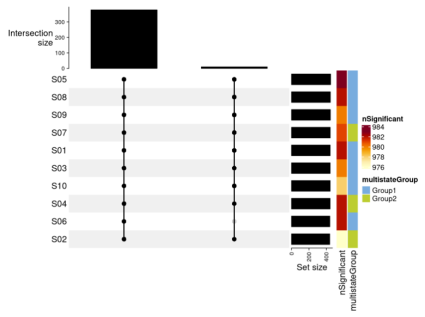
通过这些可视化工具,我们可以更直观地理解不同生物学状态下基因表达的模式和共享性,从而为精准医学和个性化治疗提供更深入的洞见。
五、多状态QTL模式的特征分析
- 多状态QTL测试的分类
在进行多状态测试校正后,我们通常希望识别出全局的、多状态的和独特的QTL。plotCompareStates函数可以返回一个包含ggplot2对象的列表,以及一个表格,分别作为第一个和第二个元素。这些元素可以通过名称“plot”或“counts”来访问。
sim_top <- runTestMetrics(sim_top)
plotCompareStates(sim_top, x=”S01″, y=”S02″)
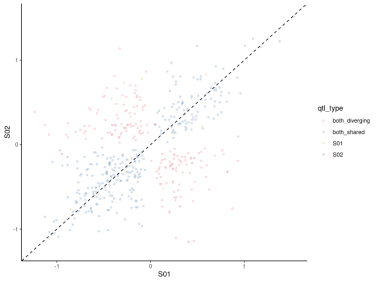
这段代码将生成一个图表,显示不同状态下QTL的分布情况。我们可以通过查看“both_diverging”(双方都在发散)、“both_shared”(双方都共享)以及特定状态下的QTL数量。
# 输出QTL类型的计数结果
table(rowData(sim_top)$qtl_type)
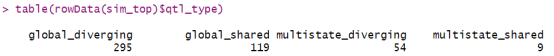
这将给我们一个不同类型QTL的概览,例如全局发散型、全局共享型和多状态发散型QTL的数量。
# 显示显著QTL的数量分布
hist(rowData(sim_top)$nSignificant)
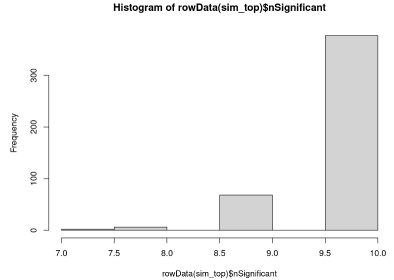
2. 可视化多状态QTL
plotQTLClusters函数可以用来生成每个状态下eQTL beta值的热图。每一行代表一个SNP-基因对,而列代表不同的状态。可以通过指定rowData或colData中的列名来添加行和列的注释。
sim_top_ms <- subset(sim_top, qtl_type_simple == “multistate”)
plotQTLClusters(
sim_top_ms,
annotateColsBy=c(“multistateGroup”),
annotateRowsBy=c(“qtl_type”, “mean_beta”, “QTL”))
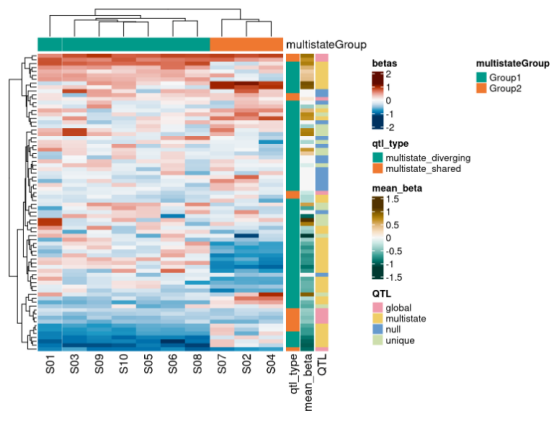
这段代码将生成一个热图,显示多状态QTL在不同状态下的效应大小。通过这种可视化方式,我们可以直观地观察到不同状态下QTL效应的变化,以及它们是如何受到不同生物学条件影响的。
通过使用multistateQTL包,我们现在可以更全面地理解基因如何在不同状态下影响复杂的数量性状,为未来的精准医疗和个性化治疗提供了强有力的数据支持。如果你对如何应用这个强大的工具有任何疑问,记得找小师妹,让我们一起探索基因组的奥秘!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~