嘿科研小伙伴们!你是否曾在研究蛋白质水解过程中感到迷茫?是否在寻找一种方法来分析那些复杂的酶切数据?别担心,小师妹今天就要给大家带来一个激动人心的生物信息学工具——proteasy包。
在这个领域里,蛋白酶(protease)的切割作用对许多重要的生理过程都有着深远的影响,而proteasy包正是为了帮助我们更好地理解和分析这些过程而设计的。它能够让我们轻松地从MEROPS数据库中提取信息,识别特定底物的潜在酶切位点,并对这些位点进行详细的分析。
现在,就让我们一起来学习如何使用proteasy包,让它成为我们研究过程中的得力助手。注意哦,这个R包操作占用内存比较大,建议使用服务器哦,欢迎联系小师妹租赁性价比高的服务器~
准备好了吗?让我们开始这段精彩的探索之旅,一起揭开蛋白质酶切的神秘面纱!如果有任何疑问,小师妹随时在这里为你解答哦!
首先,让我们安装和加载proteasy包:
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“proteasy”)
library(“proteasy”)
一、使用方法
1. 根据底物查找蛋白酶
想要快速识别可能切割特定底物的蛋白酶吗?小师妹来教你用`searchSubstrate`函数。这个函数能帮我们从MEROPS数据库中检索记录的蛋白酶。
简单两步,轻松查找:
(1) 获取蛋白酶列表
设置`summarize = TRUE`,函数会返回一个列表,包含所有已知能切割特定底物的、经过审核的蛋白酶。
searchSubstrate(protein = “P01042”, summarize = TRUE)

(2) 获取详细切割事件
设置`summarize = FALSE`,函数会返回一个详细的数据表,展示每个切割事件的信息。
searchSubstrate(protein = “P01042”, summarize = FALSE) |> head()

处理多个底物?没问题!
没错,这个函数还能同时处理多个底物,帮你一次性获取所有相关蛋白酶的信息。
searchSubstrate(protein = c(“P01042”, “P02461”), summarize = FALSE) |> tail()
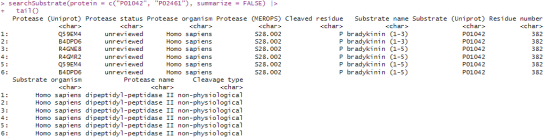
通过这种方式,我们不仅能够了解哪些蛋白酶可能参与了特定底物的切割,还能深入了解每一次切割事件的具体细节。这对于研究蛋白酶的功能和它们在生物体内的调控机制来说,是非常有价值的信息。
- 根据蛋白酶查找底物
如果你想知道某个特定的蛋白酶能够切割哪些底物,`searchProtease`函数就是为你准备的。这个函数能够帮你找出特定蛋白酶的所有已知切割底物。下面,我们将以基质金属蛋白酶-12(MMP-12,P39900)为例,演示如何使用这个函数。
只需设置`summarize = TRUE`,函数就会返回一个向量,列出MMP-12能够切割的所有底物。
searchProtease(protein = “P39900”, summarize = TRUE)

如果你想要更详细的信息,比如每次切割的具体位置和类型,只需将`summarize`设置为`FALSE`,函数就会返回一个包含这些详细信息的数据表。
searchProtease(protein = “P39900”, summarize = FALSE) |> head()

通过这个功能,我们可以深入了解特定蛋白酶如何影响各种底物,这对于研究蛋白酶的功能和它们在疾病中的作用尤为重要。小师妹提醒你,这个工具非常实用,无论你是研究特定蛋白酶还是探索蛋白酶的潜在底物,都能派上大用场哦!
- 为切割的肽段寻找可能的蛋白酶
当你手头有一堆切割后的肽段,而你想知道哪些蛋白酶可能是“幕后黑手”时,`findProtease`函数就能大显身手了。这个函数会自动将肽段序列映射到完整的蛋白序列上,并获取肽段的起始和终止位置。然后,它会在MEROPS数据库中搜索这些位置,返回所有匹配的蛋白酶。
步骤一:准备数据
首先,我们需要准备一个包含UniProt ID和相应肽段序列的向量。
protein <- c(“P02671”, “P02671”, “P68871”, “P01011”, “P68133”, “P02461”, “P0DJI8”, “P0DJI8”, “P0DJI8”)
peptide <- c(“FEEVSGNVSPGTR”, “FVSETESR”, “LLVVYPW”, “ITLLSAL”, “DSYVGDEAQS”, “AGGFAPYYG”, “FFSFLGEAFDGAR”, “EANYIGSDKY”, “GGVWAAEAISDAR”)
# 或者直接加载小师妹准备的数据哦~
#protein1 <- read.csv(“protein.csv”, header = TRUE, stringsAsFactors = FALSE)[,1]
#peptide1 <- read.csv(“peptide.csv”, header = TRUE, stringsAsFactors = FALSE)[,1]
步骤二:自动映射和搜索
使用`findProtease`函数,如果未指定肽段的起始和终止位置,函数将自动通过匹配完整蛋白序列来确定这些值。
res <- findProtease(protein = protein, peptide = peptide, organism = “Homo sapiens”)
步骤三:查看结果
我们可以用三种方式查看结果:
substrates(res) |> head() # 显示输入序列的数据

proteases(res) |> head() # 显示可能切割该蛋白的已知蛋白酶

cleavages(res) |> head() # 显示相关的蛋白水解事件的详细信息

除了上面三个步骤,我们还可以进行一些额外分析:
# 了解经过审核的蛋白酶在N端和C端的切割事件比例:
cl <- cleavages(res)[`Protease status` == “reviewed”]
cl$`Cleaved terminus` |> table() |> prop.table() |> round(digits = 2)

# 检查切割的氨基酸分布情况:
cl$`Cleaved residue` |> table() |> barplot(cex.names = 2)

# 查找涉及最多切割事件的蛋白酶:
cl[!duplicated(Peptide), .(count = .N), by = `Protease (Uniprot)`]
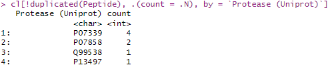
注意哦,如果你指定了起始和终止位置,函数将不会自动查找指定蛋白/肽段的序列数据,这意味着底物的序列细节将不可用。
cl_by_pos <- findProtease(
protein = “P02671”,
peptide = “FEEVSGNVSPGTR”,
start_pos = 413,
end_pos = 425)
substrates(cl_by_pos)

通过这种方式,我们可以深入了解蛋白酶如何作用于特定的肽段,为研究蛋白酶的功能和调控机制提供了有力的工具。
- 在MEROPS中查找蛋白酶详情
如果你想要直接在MEROPS数据库中查看某个蛋白酶的详细信息,可以使用`browseProtease`函数。这个函数接受一个UniProt或MEROPS的ID,并在网页浏览器中打开对应的MEROPS摘要页面。
browseProtease(“P07339”, keytype = “UniprotID”) # (打开网页浏览器)
在MEROPS数据库中指定蛋白酶的详细页面,我们可以找到关于该蛋白酶的更多数据,包括它的分类、底物、抑制剂以及相关的文献信息。小师妹提醒你,虽然这个函数很方便,但记得在有网络连接的情况下使用哦!
- 应用案例
- 可视化蛋白酶作用网络
接下来,我们可以用一种直观的方法来探究两种底物的蛋白酶作用模式:将它们呈现为一个蛋白-蛋白相互作用网络图,其中底物以红色节点表示,相关的蛋白酶则以蓝色节点显示。
首先,我们需要加载一些必要的R包,并定义我们想要研究的底物列表。
library(igraph)
library(data.table)
# 定义研究的底物
proteins <- c(‘P01011′,’P02671’)
然后,我们利用`searchSubstrate`函数查找这些底物的已知蛋白酶,并筛选出状态为“reviewed”的蛋白酶。
# 查询切割这些底物的蛋白酶
res <- searchSubstrate(protein = proteins, summarize = FALSE)
# 筛选状态为“reviewed”的蛋白酶
res <- res[`Protease status` == “reviewed”]
为了构建网络图,我们提取了与蛋白酶对底物的切割作用相关的两列数据。
# 提取构建网络所需的数据
res <- res[, c(“Protease (Uniprot)”, “Substrate (Uniprot)”, “Cleavage type”)]
使用igraph包,我们根据这些数据构建了一个有向无环图(DAG)。
# 构建网络图
g <- igraph::graph_from_data_frame(res,
directed = TRUE,
vertices = unique(
c(res$`Protease (Uniprot)`,
res$`Substrate (Uniprot)`)))
最后,我们绘制这个网络图,并对其进行美化,以便于我们能更清楚地看到每个蛋白酶和底物之间的相互作用。
# 设置随机种子以复现结果
set.seed(104)
# 绘制网络图
plot(g,
vertex.label.family = “Helvetica”,
vertex.size = 14,
vertex.color = ifelse(V(g)$name %in% res$`Protease (Uniprot)`,
“#377EB8”, “#E41A1C”),
vertex.label.cex = 1,
vertex.label.color = “white”,
edge.arrow.size = .6,
edge.color = ifelse(E(g)$`Cleavage type` == “physiological”,
“#01665E”, “#8E0152”),
layout = igraph::layout.davidson.harel)
# 添加图例
legend(title = “Nodes”, cex = 2, horiz = FALSE,
title.adj = 0.0, inset = c(0.0, 0.2),
“bottomleft”, bty = “n”,
legend = c(“Protease”, “Substrate”),
fill = c(“#377EB8”, “#E41A1C”), border = NA)
legend(title = “Edges”, cex = 2, horiz = FALSE,
title.adj = 0.0, inset = c(0.0, 0.0),
“bottomleft”, bty = “n”,
legend = c(“Physiological”, “Non-physiological”),
fill = c(“#01665E”, “#8E0152”), border = NA)

通过这种网络图,我们可以直观地观察到特定蛋白酶与底物之间的潜在互作关系,这对于理解蛋白质在生物体内的功能和调控网络非常有帮助。
- 序列相似性热图
proteasy包能够自动根据蛋白ID查找蛋白序列,这得益于ensembldb和Rcpi这两个库的支持。我们可以使用 `substrates()` 函数来获取这些序列。在这里,我们将研究一些蛋白和肽段水平上的序列相似性矩阵,并将它们作为热图绘制出来。为了进一步分析,我们还会用切割数据对这些热图进行注释。
首先,我们需要加载一些额外的库,并准备输入数据:蛋白和相关的肽段序列。
library(Rcpi)
library(viridis)
suppressPackageStartupMessages(library(ComplexHeatmap))
接着,我们可以定义我们感兴趣的蛋白列表和相关的肽段序列,这些将作为我们分析的起点。
protein <- c(‘P01011′,’P01011′,’P01034′,’P01034’,
‘P01042′,’P02671′,’P02671′,’P68871’,
‘P68871′,’P01042’)
peptide <- c(‘LVETRTIVRFNRPFLMIIVPTDTQNIFFMSKVTNPK’,’ITLLSAL’,
‘KAFCSFQIY’,’AFCSFQIY’,’DIPTNSPELEETLTHTITKL’,’FEEVSGNVSPGTR’,
‘FVSETESR’,’LLVVYPW’,’VDEVGGEALGR’,’KIYPTVNCQPLGMISLM’)
使用findProtease函数,我们可以查找与特定底物相关的蛋白酶,并获取它们的切割数据。
# 查找与上述底物相关的切割数据
res <- findProtease(protein = protein,
peptide = peptide,
organism = “Homo sapiens”)
接下来,我们可以提取底物信息,并计算了蛋白序列之间的相似性,这有助于我们理解不同蛋白之间的同源性。
# 获取底物信息
ss <- substrates(res)# 仅展示唯一的序列
ss_uniq <- ss[!duplicated(`Substrate sequence`)]
# 计算蛋白(底物)序列相似性
psimmat = Rcpi::calcParProtSeqSim(ss_uniq$`Substrate sequence`,
type = ‘global’,
submat = ‘BLOSUM62’)
rownames(psimmat) <- colnames(psimmat) <- ss_uniq$`Substrate (Uniprot)`
利用计算出的序列相似性数据,我们绘制了一个热图,以直观展示不同蛋白序列之间的相似度。
# 热图绘制
ComplexHeatmap::Heatmap(psimmat, col = viridis::mako(100))

同样的方法也应用于肽段序列,我们计算了它们的序列相似性,并用切割残基和末端信息对每一行进行了详细的注释。
# 获取切割细节
cl <- cleavages(res)
# 计算肽段序列相似性
pep_psimmat = Rcpi::calcParProtSeqSim(cl$Peptide, type = ‘global’,
submat = ‘BLOSUM62’)
# 添加注释
rsd <- cl$`Cleaved residue`
cols <- c(“#8DD3C7”, “#FFFFB3”, “#BEBADA”, “#FB8072”)
names(cols) <- unique(rsd)
ha1 <- ComplexHeatmap::columnAnnotation(`cleaved residue` = rsd,
col = list(`cleaved residue` = cols))
tn <- cl$`Cleaved terminus`
cols <- c(“#B3E2CD”, “#FDCDAC”)
names(cols) <- unique(tn)
ha2 <- ComplexHeatmap::columnAnnotation(terminus = tn,
col = list(terminus = cols))
rownames(pep_psimmat) <- cl$`Substrate (Uniprot)`
最后,我们为肽段序列绘制了另一个热图,其中包括了切割位点的详细信息,这有助于我们深入理解蛋白酶如何作用于这些特定的肽段。
# 热图绘制
ComplexHeatmap::Heatmap(
pep_psimmat,
name = “sequence\nsimilarity”,
col = viridis::mako(100),
show_column_names = FALSE,
row_names_gp = grid::gpar(fontsize = 6.5),
top_annotation = c(ha1, ha2))
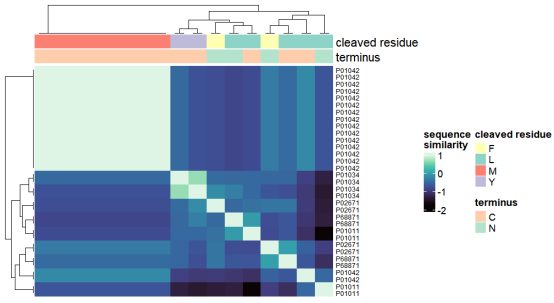
通过这些步骤,我们能够将复杂的生物信息数据转化为直观的图形表示,从而更深入地理解蛋白酶与其底物之间的相互作用。
通过使用proteasy包,我们不仅成功地分析了蛋白序列的相似性,还深入理解了蛋白酶如何精确地作用于特定肽段。这强大的工具将大大加速你在蛋白质组学研究中的探索步伐。如果你对这个过程还有任何疑问,或者需要进一步的帮助,随时可以联系小师妹哦!
无论你是在优化代码,还是在云端进行便捷的分析,云生信神器都能为你提供强大的支持。欢迎试试我们的云生信神器,只需一键上传数据,想要的图就能轻松get~